Introduction
Hereditary hemorrhagic telangiectasia (HHT), also known as Osler-Rendu-Weber disease, is an autosomally dominant inherited vascular disorder. The main characteristics of the disease are abnormal capillary connections between arterioles and venules of the skin and mucosa which present as telangiectases. Telangiectases are abnormal arteriovenous communications of capillary origin also described as focal dilatations of postcapillary venules which often cause small capillary bleedings. Apart from small blood vessels, abnormalities can also affect communication between larger arteries and veins, which can lead to life threatening arteriovenous malformations. These malformations can be found in the lungs, liver, brain and digestive system (1). Small telangiectases represent mainly cosmetic problem as opposed to arteriovenous malformations which can cause severe blood loss, hypoxemia and embolism. HHT was first described in 1865 and until recently it was considered an extremely rare disease with incidence of 1 in 100,000. However, new studies show that HHT incidence is more frequent and estimated at 1 in 5,000–8,000. The main reason for misdiagnosis is its wide variety of symptoms (1). Diagnosis of HHT is based on four main clinical features called Curacao criteria which were established in 1999 to improve and facilitate admission of individuals with HHT (2). These features are as follows: spontaneous recurrent epistaxis (nosebleeds), mucocutaneous telangiectases (abnormal capillary connections that mostly appear on the skin and mucosa), visceral arteriovenous malformations (inadequate connection between arteries and veins in the liver, lungs, digestive system and brain) and an affected first degree relative as indication of autosomal dominant inheritance. Definite diagnosis of HHT is made if three of the four mentioned criteria are present. HHT can be suspected if there are two positive criteria and, in case that only one criteria is present, HHT is considered to be unlikely (2).
The occurrence of blood vessel disorder is linked to two recently identified genes involved in angiogenesis. Protein transcripts of these genes participate significantly in recovery and regeneration process of impaired blood vessels which arise as a result of increased blood circulation, formation of thrombus, local inflammations and similar processes. The first described mutation was established on chromosome 9 in gene encoding for endoglin (ENG gene) (3). The second gene linked to HHT is located on chromosome 12 and identified as gene for activin-receptor-like kinase 1 (ACVRL1 gene) (4). According to affected gene, HHT is divided into two clinically indistinguishable forms: HHT1 caused by mutations in ENG gene and HHT2 in case of mutations inACVRL1 gene. Results of newer studies indicate two additional genes associated with HHT, MADH4 gene, mutated in a combined syndrome of juvenile polyposis and HHT, and an unidentified HHT3 gene related to chromosome 5 (5).
Considering great variety of symptoms and clinical manifestations especially in less serious cases, HHT is often misdiagnosed for one of more common diseases like iron deficiency anemia or idiopathic telangiectasia (6). Since specific genetic testing is not available in Croatia, it is of great importance to recognize individual symptoms and associate them into the unique clinical entity of HHT. Wrong diagnosis postpones appropriate therapeutic measures and, regarding the progressiveness of HHT, increases the possibility of chronic complications which can remain unrecognized till advanced stages of the disease.
This report describes a case of a female patient who has been diagnosed with HHT after she was unsuccessfully treated with misdiagnosis for approximately ten years. Major goal of this case report was not only to remind readers of this rare chronic disease but also to point out many complications that may accompany it, and describe how to correctly interpret laboratory test results.
Case report
A female patient, born in 1955, has been treated for hereditary hemorrhagic telangiectasia (Osler-Rendu-Weber disease) in the Outpatient Division of Hematology, Department of Internal Medicine, Rijeka Clinical Hospital Center. Physical examination revealed signs of severe anemia: fatigue and weariness, weak and sparse hair, extremely pale skin and mucosa. Capillary telangiectases were also present, mostly on lips and nose. The patient suffered from massive nosebleeds on daily basis and also on few occasions from bleedings in the larynx. The patient was very slim and case history revealed slow but constant body weight loss.
When she presented for her regular follow-up in the outpatient clinic, her hemoglobin levels varied between 26 g/L and 60 g/L, erythrocyte count was around 2.5 × 1012/L, hematocrit 0.210 L/L and MCV(mean corpuscular volume) 75 fL. After blood transfusion, hemoglobin levels rose up to 90 g/L at most and erythrocyte count to 3.1×1012/L. Due to frequent blood transfusions, RDW (red blood cell distribution width) was around 20%, which indicated significant anisocytosis resulting from the presence of different erythrocyte populations. Although oral and intravenous iron therapy was applied regularly, serum iron concentration was invariably low (1 μmol/L) accompanied by high levels of unsaturated serum iron binding capacity (UIBC) (60 µmol/L), normal total serum iron binding capacity levels (TIBC) (61 μmol/L) and ferritin levels nearing the lower reference interval value (10 μg/L).
Since 2005, the patient has been receiving transfusions of deplasmatized blood monthly with previous corticosteroid treatment which prevents possible immune reaction against donor’s blood antigens. When needed, the patient is also given human recombinant erythropoietin and iron therapy as well as multivitamins and histamine H2 receptor antagonist for protection of gastric mucosa, and required to rest and present for regular checkups.
One of the criteria for HHT diagnosis is presence of the disease in first-degree relatives. It was not possible to determine the disease in the patient’s parents as they died before she was correctly diagnosed. Family history revealed that the patient’s father died of pulmonary tuberculosis. The patient’s mother was anemic and often tired, which could indicate anemia caused by hereditary hemorrhagic telangiectasia. The patient’s brother and sister show similar symptoms but in a milder form. As opposed to sister who does not need blood transfusions, her brother occasionally receives blood transfusions.
Case history
Regarding late determination of the diagnosis, appearance of symptoms and complications and therapeutic approach, the course of disease could be divided into four time periods as presented in Table 1. During development of the disease and therapy, the patient showed some non-typical signs apart from findings typical for HHT.
Table 1. Disease course overview according to blood hemoglobin concentration, erythrocyte count, hematocrit, serum iron concentration and deplasmatized blood transfusions. Symbol interpretation: N – value within reference interval, ¯ – low value according to reference interval.
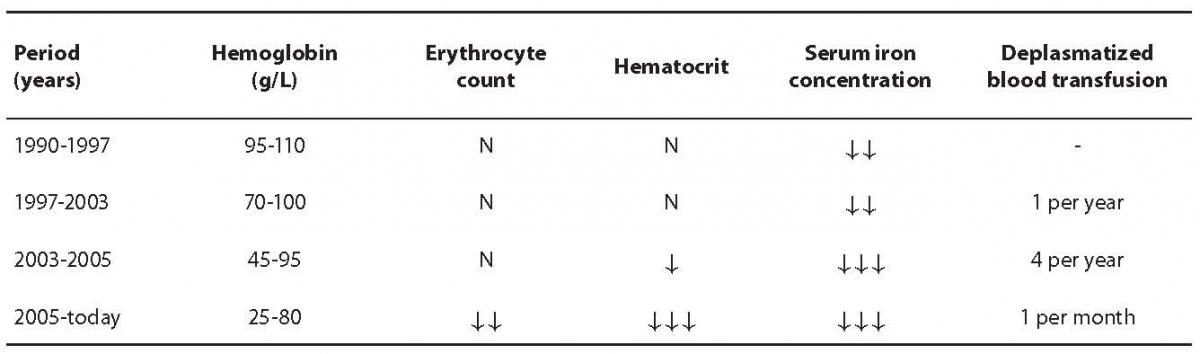
Due to symptoms of anemia in 1990, the patient was referred to the Outpatient Division of Hematology, Department of Internal Medicine, Rijeka Clinical Hospital Center. According to performed laboratory tests, the patient was diagnosed with severe iron deficiency anemia and started with iron therapy. In scarce medical documentation and anamnesis from that time, the patient was known to have had tuberculosis in 1986. She was treated and monitored for years with the diagnosis of iron deficiency anemia, and was taking iron substitutes during that period. However, this therapy did not improve iron status or diagnosed anemia.
She was again thoroughly examined seven years later. The examination revealed loss of body weight, occasional nosebleeds and fewer menstrual bleedings. Pulmonary examination showed inactive tuberculosis, and gynecological report indicated no abnormalities. As opposed to irigography and rectoscopy findings which were normal, esophagogastroduodenoscopy revealed well restricted petechial bleeding in several parts of the stomach. Test for occult bleeding was positive from three consecutive stool samples. In 2000, she was found to have vocal cord polyps and was referred to the Zagreb Clinical Hospital Center for surgery. During preoperative examination, diagnosis of severe iron deficiency anemia was questioned and shortly afterwards the diagnosis of HHT or Osler-Rendu-Weber disease was made according to diagnostic criteria. In that period (1997 – 2003) the patient presented for her regular follow-ups and was checked for occult bleeding which was negative. The condition of the digestive tract was monitored using colonoscopy and esophagogastroduodenoscopy. However, these imaging tests, apart from expansion of petechial bleeding from stomach to duodenum, did not reveal any changes in the patient’s condition or the progress of the disease during that time.
During the period stated above, hemoglobin concentration varied from 70 g/L to 100 g/L and erythrocyte count was normal (about 3.5 × 1012/L), relative reticulocyte percentage exceeded the upper reference interval limit (3.8%) and MCV was low, around 67 fL. Serum iron concentrations showed values from 0.7 µmol/L to 11.3 µmol/L, UIBC level was high, with a constant TIBC level. Results of laboratory coagulation and hemostasis tests were within reference intervals during the entire period of observation. Some parameters varied depending on iron replacement therapy and deplasmatized blood transfusions which were during that period administered once a year.
The period in the course of disease that followed was marked with severe general status. In 2003 patient was hospitalized at the Division of Hematology, Department of Internal Medicine, Rijeka Clinical Hospital Center. She experienced massive epistaxis on a daily basis. After discharge from hospital, the patient was monitored more often than before, approximately once every three months, and during each checkup she was given deplasmatized blood transfusions after previous corticosteroid treatment. On arrival to the outpatient treatment, the patient’s hemoglobin levels were around 45 g/L and around 95 g/L after transfusion of deplasmatized blood. Although erythrocyte count was within the reference interval, hematocrit values were low, usually around 0.250 L/L. Serum iron concentration was around 1.5 µmol/L, TIBC level was near the upper reference interval value, 73 µmol/L. At that time, the patient’s ferritin levels were found to be low (7 µg/L). Positive occult blood test indicated bleeding in digestive tract and coagulation test values were still within normal range. During examination the patient was subjected to imaging tests. Multislice computed tomography (MSCT) of the brain showed restricted areas of vascular pathological structures that could be associated to capillary telangiectases which were consistent with diagnosis. MSCT of the chest revealed inflammation residues, probably pulmonary tuberculosis. The examination also showed a shunt between a branch of the pulmonary artery and pulmonary vein in the right middle lung lobe, which indicated the presence of arteriovenous malformation characteristic for HHT diagnosis.
The features that marked the latest period in the course of disease (since 2005) are extremely low hemoglobin levels and regular deplasmatized blood transfusions as described previously.
Discussion
Low blood hemoglobin concentration should always be considered with great attention when evaluating laboratory test results. Hemoglobin concentration of 26 g/L may well imply a preanalytical error such as incorrect specimen collection, sample dilution with intravenous infusion liquid or the presence of microclots which prevented accurate analysis. However, with the preanalytical error excluded, low hemoglobin concentrations can be expected not only in patients with massive bleeding and acute threat to vital functions but also in patients with chronic diseases like the one described above, i.e. in a patient with hereditary hemorrhagic telangiectasia.
Limitations of this case report arise from scarce medical documentation. As the patient was treated in several medical institutions during a long period and her medical documentation was not complete, some test results could not be traced.
Severe anemia, like the one present in the reported patient, is not a characteristical finding in HHT and until now only 20 similar cases have been reported in literature (7). Anemia is most often the consequence of chronic gastrointestinal bleeding and in some cases of massive epistaxis (7). The reported patient experienced her first epistaxis around the age of 30, somewhat later than specified in the literature which describes epistaxis as a first clinical manifestation of the disease in more than 90% of cases that occurs before the age of 20 (8). However, our patient’s nosebleeds became over the years ever more intense and frequent so that nowadays they occur on a daily basis. The case history states bleedings in the larynx on a few occasions, while esophagogastroduodenoscopy, apart from petechial bleeding in the stomach and duodenum, did not reveal with certainty any significant bleeding typical for HHT.
Although occult blood test showed to be occasionally positive, patients who suffer from heavy epistaxes usually swallow considerably large amounts of blood which can result in positive fecal occult blood testing (9). Chronic, life threatening anemia in the reported patient, with extremely low concentrations of serum iron and ferritin, are probably due to recurrent nosebleeds. Unlike serum iron and ferritin concentrations, which are consistent with published data, total serum iron binding capacity values were not as high as expected (10). Results of laboratory hemostasis tests as well as the thrombocyte count were normal during the whole period of the patient’s follow-up. These findings are in accordance with published data which indicate that HHT patients have unimpaired coagulation and normal thrombocyte function (11).
Besides mucocutaneous telangiectases, HHT is also manifested through formation of a direct communication between larger arteries and veins which are called arteriovenous malformations. These arteriovenous malformations mainly occur in the lungs, liver, brain and digestive tract (12). MSCT of the patient’s chest detected pulmonary arteriovenous malformation, yet without any pulmonary symptoms. The most common visceral manifestation of HHT is pulmonary arteriovenous malformation (30%). It is also estimated that 60–70% of all pulmonary arteriovenous malformations develop in patients with HHT (13). Patients are often asymptomatic and usually experience only dyspnea and weariness which can also be due to anemia. Besides arteriovenous malformations, MSCT of the chest revealed residues of pulmonary tuberculosis. Association between pulmonary tuberculosis and arteriovenous malformations has been poorly described in literature but some cases have been reported (14,15). There is evidence of some misdiagnosed tuberculosis that was only later discovered to be an arteriovenous malformation within HHT (16). One of the features of tuberculosis is intensive angiogenesis in inflammatory mass occurring in order to increase blood supply. As new blood vessels are of poor quality, they are likely to give rise to arteriovenous malformations (14). In case of the described patient there is a possibility that pulmonary tuberculosis, as an inflammatory process, stimulated the development of arteriovenous malformation. On the other hand, capillary abnormalities which were found in our patient can be the basis for easy onset of infection such as tuberculosis against which an otherwise healthy organism would defend. Imaging procedures like MSCT cannot determine the exact order of pathological events in the lungs.
MSCT of the patient’s brain showed minor vascular abnormality, which is consistent with the diagnosis even though cerebral vascular malformations are associated with HHT in low percentage (< 10%) of patients (17). Vascular cerebral abnormalities can lead to acute complications such as ischemic attack or brain abscess. Apart from vascular causes, complications often arise as a result of arteriovenous shunts in the lungs, the main reason of deficient capillary blood filtration that facilitates formation of a thrombus. When in system circulation, the thrombus can obstruct blood vessels of vital organs and, even in the presence of the mildest pulmonary symptoms, patients can experience acute neurological complications (17,18).
As HHT is a genetic disorder, therapeutic efforts remain symptomatic. The only effective treatment would probably be gene therapy which is presently not available. HHT centers in health systems of developed countries support treatment and monitoring of the disease and also provide genetic testing for mutations that have been proven to be connected with HHT (3-5). According to current possibilities, treatment of the disease is based on the management of acute nasal bleeding and preventive measures. Unfortunately, therapeutic aproaches to nosebleeds are often short-termed. Besides management of epistaxes, HHT patients have constant need for blood transfusions which depend on bleeding intensity. At the beginning of treatment, the reported patient was given blood transfusions once a year and during a ten year period she developed the need for blood transfusions on monthly basis together with human recombinant erythropoietin. HHT patients also take iron substitutes whose dosage depends on the severity of sideropenia.
Hereditary hemorrhagic telangiectasia is a progressive and extremely debilitating disease. Patients are also exhausted by long lasting treatments, constant monitoring and often wrong initial diagnosis. By reporting this case we wanted to draw attention to the possibility of the rare but chronic disorder that can have significant effect on laboratory test results which in that case might be the first sign of the disease. Also, our intention was to remind of the importance of careful diagnosing and medical decision making, particularly in the case of anemia due to its considerable variety and complexity.