Introduction
Neonatal cholestasis is defined as obstruction of bile flow mostly caused by structural or functional anomaly of hepatobiliary tract. Neonatal cholestasis could be intrahepatic or extrahepatic. Infants with cholestasis present with jaundice, dark urine, acholic stools and hepatomegaly (1). Intrahepatic disorder associated with cholestasis in a newborn is, among others, α1-antitrypsin (AAT) deficiency, one of the most common hereditary liver diseases leading to liver transplantation in children (2).
AAT is a glycoprotein, inhibitor of serine proteases (elastase, cathepsin G and proteinase 3) that is mostly secreted by hepatocytes and to a lesser extent by alveolar macrophages and monocytes (3). The AAT gene located on chromosome 14 is very polymorphic with over 100 mutations already described (4).
The AAT phenotype variants are categorized by their electrophoretic mobility and serum concentrations to protease inhibitor (Pi) system. The most common, normal variant is middle migrating PiM allele characterized by normal AAT serum concentrations. The most frequent deficient variants are PiZ and PiS, characterized by reduced AAT serum concentrations and slow (S) or very slow (Z) migration velocity (5).
PiZZ and PiSS homozygous genotypes reduce AAT serum level to 15% and 60% of normal serum concentrations, respectively. PiSZ phenotype shows reduction of the AAT serum concentration to 30–35% of the normal value (6).
AAT deficiency could clinically present as pulmonary emphysema, chronic bronchitis and asthma in elderly, but infants have other symptoms. In infancy and childhood, AAT deficiency can cause prolonged obstructive jaundice with severe liver dysfunction leading to lethal cirrhosis in 1–2% of deficient infants (7-9).
Intriguingly, only 10–15% PiZZ individuals develop clinical cholestasis in childhood with diverse subsequent course (10).
In this report, we present a case of a newborn with severe intrahepatic cholestasis associated with α1-antitrypsin deficiency.
Case report
A 15-day-old male newborn (body length 0.61 m, body weight 5.1 kg) was admitted to the Gastroenterology ward of the Pediatrics Department, Sestre milosrdnice University Hospital, due to pale stools and suspected cholestasis. The mother had noticed acholic stools when the boy was 9 days old. The jaundice was not present although the urine was dark colored. The newborn was breastfed with a commercial food supplementation.
The patient’s personal history revealed that he was born in the 38th gestational week via elective Cesarean section as a third-born child. His birth weight was 2580 g and birth length 48 cm. After 7 days he was released from the maternity hospital with the total weight of 2390 g. The newborn’s parents and two sisters were healthy.
Physical examination on admission to the Pediatrics Department showed no particularities, with stable general condition. Biochemical, hematological and coagulation tests, as well as ultrasound examination and Technetium 99m-hepatic imino diacetic acid (HIDA) scintigraphy were performed for the exclusion of bile duct anomalies.
The general biochemical parameters determined were: total and conjugated bilirubin, AST, ALT, GGT, ALP, 5’-NU, bile acids, α1-antitrypsin, α-fetoprotein, total cholesterol, LDL and HDL cholesterol, triglycerides, CRP, IgG, IgA and IgM immunoglobulins in serum. Erythrocyte sedimentation rate (ESR), differential blood count, bleeding time, clotting time, prothrombin time (PT) and fibrinogen were also determined. Table 1 shows only the values which were not in the reference ranges while other measured parameters were normal.
Table 1. Biochemical parameters measured in the infant aged 15 days and 4 months.
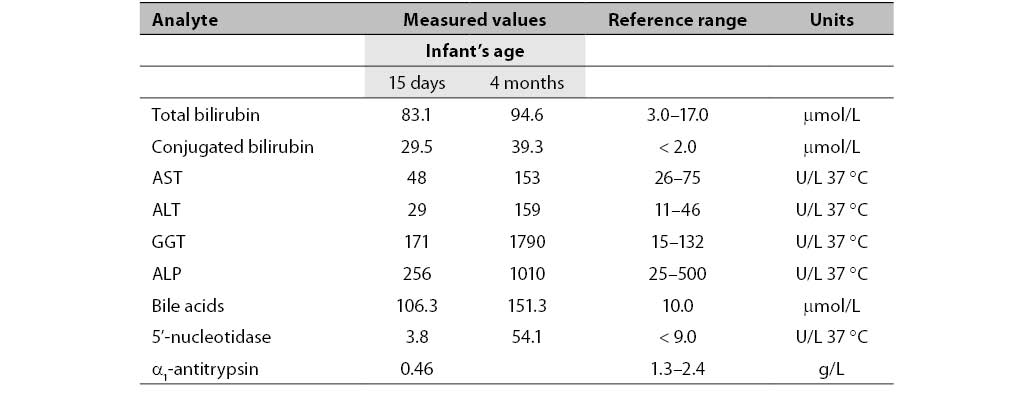
Concentrations of total and conjugated bilirubin were increased already in the 15-day-old newborn. GGT activity was also increased. Activities of other liver enzymes (AST, ALT and ALP) were normal. The concentration of the newborn's bile acids was extremely increased. Due to decreased AAT serum concentration, genotyping and phenotyping were performed (11, 12) and revealed AAT deficiency in the PiZZ genotype. Due to increased concentration of unconjugated bilirubin in the newborn’s serum, the genotyping for Gilbert’s disease was performed but the polymorphism was not found.
Two HIDA abdomen scintigrams were done 24 hours after initiation of radioactive substance. The second one was done to confirm first results. In both cases the radioactive substance was normally accumulated in the liver, but without passage to the intestine. These examinations confirmed suspicion of severe intrahepatic cholestasis.
The newborn was released from the hospital after initiation of therapy for cholestatic liver disease. Four months later he was admitted again due to no signs of improvement but with exacerbation of condition. Activities of all liver enzymes (GGT, AST, ALT and ALP) were significantly above the upper reference limit.
The liver and spleen ultrasound examination showed no particularities and the size of the liver was normal.
After decreased AAT serum concentration was revealed in the newborn, AAT concentrations were also measured in the newborn’s parents, with results of 1.01 g/L for mother and 1.50 g/L for father (reference range 1.30–3.00 g/L). Also, AAT genotyping and phenotyping were performed. The mother was found to have PiSZ genotype and the father had PiMZ genotype.
For the AAT genotyping, two PCRs (S PCR and Z PCR) were performed with two sets of primers. Each PCR had its own products (S and Z products). By combining those results (the same alleles in both PCRs defining genotype) the final genotypes for the infant and his parents were read. The scanned picture of the gel is shown in Figure 1.
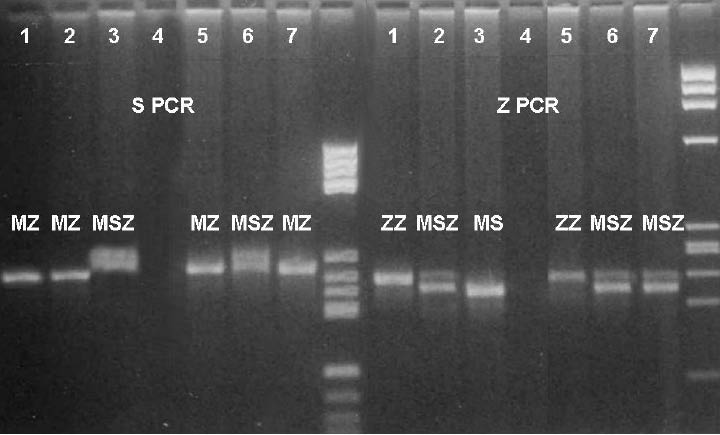
Figure 1. AAT genotyping results of S- and Z-PCR
Legend:
1 – ZZ control 5 – newborn – ZZ
2 – MZ control 6 – mother – SZ
3 – MM control 7 – father – MZ
4 – distilled water M – molecular weight marker
Discussion
The above presented case has been the most severe neonatal case of intrahepatic cholestasis caused by AAT deficiency in our hospital. In the period since 1977, approximately 30 cases of homozygous and heterozygous AAT deficiency have been observed in our clinical hospital. The first case of AAT deficiency in the hospital was described in 1977 (13). A few years ago, studies about genotyping of AAT in Croatian families were published, also from our hospital (14, 15).
The newborn's AAT serum concentration was decreased to 30–35% of the reference value, which is a higher level than expected in PiZZ genotype. The reason could be the acute inflammatory response which causes a rise in the AAT concentration.
Cases of children that died at 7 years of age due to cirrhosis caused by AAT deficiency have already been reported (16) but in our hospital this is the first case of such a severe presentation of AAT deficiency. The child presented in this case report is currently affected by liver damage and needs a liver transplantation.
An interesting finding in this case was the mother's PiSZ genotype. During 7 years of genotyping and 11 years of phenotyping, this was the first time in 152 cases (0.66 %) with suspected AAT deficiency that we found PiSZ genotype.
This case of cholestasis with jaundice is a very rare occurrence and only 6% of infants develop such clinical disease. In approximately 15% of patients, cholestatic jaundice progresses to juvenile cirrhosis (17).
Conclusion
Severe intrahepatic cholestasis associated with alpha-1-antitrypsin deficiency was confirmed in a newborn. Genotyping and phenotyping of the newborn and his parents revealed PiZZ genotype in the newborn, PiSZ genotype in his mother and PiMZ in his father. PiSZ genotype is a very rare AAT genotype which was detected in our 7-years of genotyping and 11 years of phenotyping for the first time.