Introduction
Detection and quantification of hemoglobin (Hb) fractions is clinically relevant in two situations. First, quantification of HbA1c, the major glycated form of HbA, is useful in the clinical management of diabetes. Methods for HbA1c analysis include immunoassay or chromatography. Second, the quantification of HbA2 and HbF, and the detection and quantification of Hb variants is an essential tool in the diagnosis of hemoglobinopathies (e.g., thalassemia or sickle cell syndromes).
β-thalassemia is an inherited form of anemia resulting from defective synthesis of β-globin chain. HbA2 is a critical test to identify β-thalassemia carriers because the increase in this Hb fraction is the most important diagnostic characteristic of heterozygous β-thalassemia.
HbA2 was identified more than 50 years ago as the second Hb in the blood of healthy adults (1), representing about 3% of total Hb. Later studies demonstrating HbA2 to be increased in thalassemia trait (2) and to comprise 2 α and 2 δ globin chains, led Ingram and Stretton to postulate that, because β globin synthesis was decreased, an increase of HbA2 was likely in β-thalassemia (3). Such an increase is not specific because elevated concentrations have also been reported in other conditions (4-6). In conjunction with erythrocyte indices, however, HbA2 remains a standard biomarker for β-thalassemia screening in adults.
Thalassemia syndromes are among the most common genetic disorders worldwide, with 1.7% of the world’s population carrying thalassemic genes (7). Thalassemia is prevalent in some parts of the world (Mediterranean region, up to 8%; Middle East countries, up to 10%; India, 3%–15%; and Southeast Asia, up to 9%) where it poses a major public health problem. However, non-endemic countries such as northern Europe and North America are also involved in thalassemia related problems as the result of demographic changes caused by migration of ethnic minority groups with a high frequency of thalassemic mutations (8). Data from recent epidemiological surveys indicate that in Europe there are approximately 15,000 subjects with transfusion dependent thalassemia major (9-11). In this regard, HbA2 determination plays a key role in screening programs for β-thalassemia, since an increase in this Hb fraction allows for heterozygotes to be recognized. However, some diagnostic difficulties may arise on identification of atypical carriers having slightly increased HbA2 values. The finding of such borderline subjects is not rare because of the great heterogeneity of β-thalassemia genotypes or the possible coexistence of other acquired or genetic conditions (i.e. iron deficiency, δ-thalassemia) that may mask the diagnosis of heterozygous β-thalassemia (12-14). Therefore, owing to the narrow separation between normal and pathological HbA2 values, strict analytical quality of HbA2 measurement is an essential requirement for accurate diagnosis, particularly for genetic counseling when couples at risk have to be identified.
The methods for hemoglobinopathy investigation traditionally include alkaline and acid Hb electrophoresis in combination with HbA2 quantification by electrophoresis or chromatography, and HbF quantification with these methods or alkali denaturation. Above mentioned methods are manual and time consuming (15). In the last decades, automated techniques have been developed for Hb fractionation, producing results required by clinicians, while improving laboratory efficiency and quality of results.
Cation exchange high performance liquid chromatography (HPLC) is a fully automated system for qualitative and quantitative Hb fraction analysis, suitable for laboratories with a high workload (16,17).
Samples are injected into a chromatograph equipped with an ion-exchange column that has been equilibrated with respect to pH and ionic strength. Separation of Hb species is accomplished through the use of a gradient between two mobile phases with differences in salt concentration and pH. Physical characteristics such as surface charge and the presence of hydrophilic and hydrophobic groups determine the rate of migration of each species. HPLC methods have considerable advantages over labor-intensive conventional techniques in terms of precise quantification, time savings and full automation. Currently, HPLC techniques are the most reliable methods for separation and quantification of Hb fractions, and are useful tools in presumptive identification of Hb variants but having potential limits of false interpretation in the presence of an abnormal Hb.
Materials and methods
Primus Ultra2 HPLC system
Primus Ultra2 analyzer (Primus Corporation, Kansas City, Kansas, USA) employs principles of ion exchange chromatography HPLC and spectrophotometric detection. The system has been designed to provide the maximum amount of information in the shortest time. The method combines the sensitivity and specificity of HPLC with automation software and provides assistance in data evaluation.
Samples are run in batches and every run starts with a FASC control as a retention time marker for the known hemoglobins that it contains: HbF, HbA0, HbS and HbC, as stated by the name FASC (Figures 1 and 2). FASC control ensures optimal system performance. Both whole blood and hemolysates are suitable samples.

Figure 1. Primus Ultra2 chromatogram, FASC control Quick Scan assay. Quick Scan assay: for screening patient samples. Analysis time is 4 minutes; the system quantifies HbF, HbA2, HbA0 and flags abnormal peaks. FASC control is a retention time marker for the known hemoglobins it contains: HbF, HbA0, HbC and HbS.

Figure 2. Primus Ultra2 chromatogram, FASC control high resolution assay. High resolution assay: in 10.5 minutes it further resolves abnormal hemoglobins to aid in their identification.
Five µL of whole blood are automatically sampled, hemolyzed and injected onto the column during the flow of appropriately blended buffers (mobile phases 1 and 2). Upon elution from the column, sample components pass through the spectrophotometric detector, where detection occurs at a wavelength of 413 ± 2 nm.
Two analysis methods are available, i.e. Quick Scan assay for screening patient samples. Analysis time is 4 minutes; the system quantifies HbF, HbA2, HbA0 and flags abnormal peaks; and High Resolution assay: in 10.5 minutes it further resolves abnormal hemoglobins to aid in their identification. Up to 100 Hb variants can be recognized and identified with this high resolution working method. Either program may be used to quantify HbA2 and HbF if controls have been run with the batch.
As the sample is analyzed, a chromatogram is displayed on the monitor in real time. The computer produces printed reports with sample identification information, date and time, followed by a chromatogram with retention times indicated at the apex of each peak. A peak summary report is printed after the chromatogram with retention time, relative retention time, % area and comments for each peak in the chromatogram.
The aim of the study was to evaluate analytical performance and quality of results obtained with the Primus Ultra2 system for routine estimation of HbA2 and screening for β-thalassemia.
Patients
One hundred and fifty samples from apparently healthy subjects obtained from the Blood Transfusion Department were used to establish the reference range, according to the Clinical and Laboratory Standards Institute (CLSI) guidelines. Blood cell counts and biochemical iron test results were within the reference ranges.
The reliability of HbA2 measurement by Primus Ultra2 for detection of β-thalassemia was assessed by analyzing 300 samples from consecutive adult patients with a previous diagnosis of the disease (53% male and 47% female, age range 18-95, mean age 49 years). Table 1 shows analytical data of study patients and Figure 3 a β-thalassemia carrier chromatogram.
Table 1. Hematological and biochemical parameters in the reference group and in a pool of 300 β-thalassemia patients.
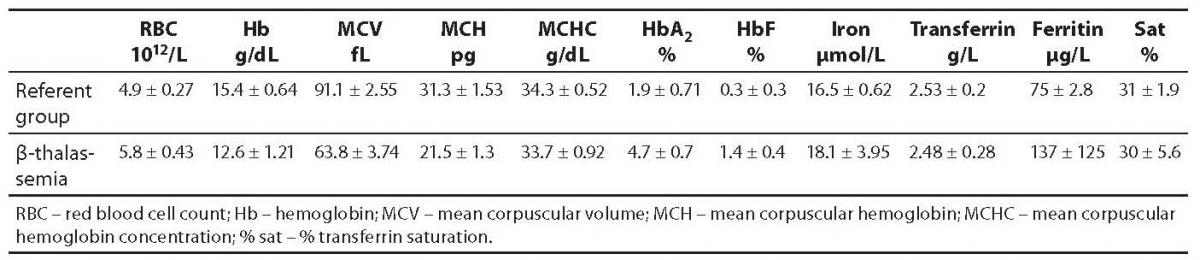

Figure 3. Primus Ultra2 chromatogram, β-thalassemia carrier. Chromatogram with retention times indicated at the apex of each peak. A peak summary report is printed after the chromatogram with the retention time, relative retention time, % area and comments for each peak in the chromatogram.
Screening for β-thalassemia is routinely performed in our laboratory by measuring red blood cell indices and level of HbA2. Molecular characterization of mutations is performed with the PCR-ASO (polymerase chain reaction-allele-specific oligonucleotide hybridization) technique.
The group of β-thalassemia carriers was diagnosed following our protocol: red blood cell indices were determined on an LH750 Beckman-Coulter (Beckman Coulter Inc. Miami, FL, USA) analyzer. Samples with erythrocytosis (red blood cell (RBC) > 5.8 x 1012/L) and microcytosis (MCV < 70 fL) were selected for HbA2 quantification, performed by HPLC HA 8160 Menarini (Menarini Diagnostics, Florence, Italy). HbA2 over the reference value of 3.5% strongly suggests β-thalassemia trait. These samples underwent molecular analysis. DNAs were purified with GenerationR capture column kit (Gentra Systems, Minneapolis, MN, USA).
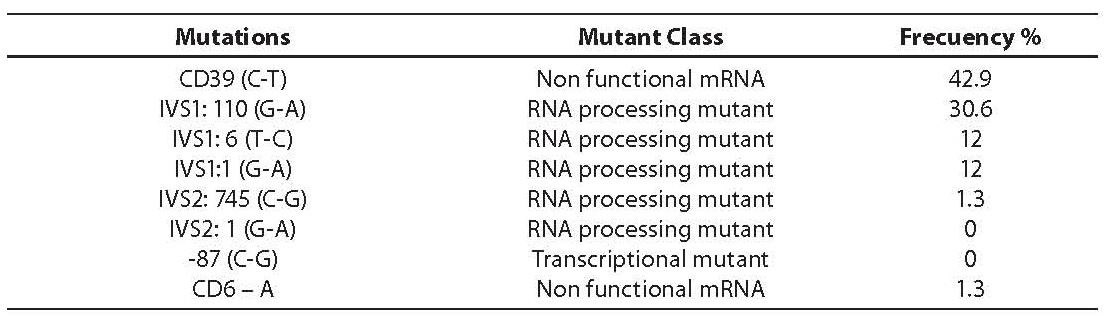
Molecular analysis for the presence of the most common Mediterranean point mutations were performed with mDxR BeTha Gene 1 (Bio Rad Laboratories, Hercules, CA, USA), based on the principle of ASO hybridization. Table 2 resumes the mutations studied, the mutant class and the results obtained for the series of 300 β-thalassemia carriers. All of them were found to be heterozygous.
Table 2. Distribution of β-thalassemia mutations expressed as percentage of the total number studied (300 β-thalassemia carriers)
Analytical evaluation
Inaccuracy
Inaccuracy was assessed by analyzing two commercially available control samples per run over 20 days. The mean, standard deviation (SD) and coefficient of variation (CV) were calculated.
Intra-assay imprecision
Two samples (one from a healthy adult and another from a β-thalassemia carrier) were analyzed 10 times within the same run. The mean, SD and CV were calculated.
Inter-assay imprecision
Two samples were assayed 3 times each day, over seven days. Samples were stored at 4 ºC every day. The mean, SD and CV of daily mean values were calculated for 7 days.
Recovery study
Recovery study by sample mixing was performed to evaluate the response of Primus Ultra2 to increasing concentrations of the analyte, i.e. HbA2.
A normal blood (2.4% HbA2) and β-thalassemia carrier sample (5.0% HbA2) were mixed at the ratios of 9:1, 8:2…1:9 and eleven samples were analyzed.
To make comparison between the theoretical expected values and measured ones, a linear regression test was performed (Method Validator 1.15, P. Marquis statistical software, Metz, France).
Results
Reference range
The mean HbA2 blood concentration was 1.9% and reference range 0.9–3.0% (mean ± 2.5 SD). All β-thalassemia carriers studied had increased HbA2 blood concentration, the values ranging from 3.3% to 6.9%, mean 4.7%.
Inaccuracy
Inaccuracy was assessed by analyzing two commercially available control samples per run over 20 days. The mean and standard deviation (SD) were calculated. The following results were obtained: 1.6% (SD 0.26) for normal control and 5.2% (SD 0.19) for high control.
Imprecision
Imprecision was tested by running samples with normal and high HbA2 concentrations. Intra-assay imprecision: two samples (one from a healthy adult and another from a β-thalassemia carrier) were analyzed 10 times within the same run. The mean and CV were calculated. Inter-assay imprecision: two samples (one from a healthy adult and another from a β-thalassemia carrier) were assayed 3 times each day, for seven days. Samples were stored at 4 ºC every day. The mean and CV of daily mean values were calculated for 7 days. Within-run CV was 1.9% for normal HbA2 level (2.2%) and 1.3% for elevated HbA2level (5.5%). Between-run CVs were slightly higher, i.e. 2.4% (normal level) and 1.7% (elevated level). Between-day CVs were 3.0% and 2.7% for normal and high HbA2 concentrations, respectively. Total CVs were 4.4% (normal level) and 3.5% (elevated level).
Recovery study
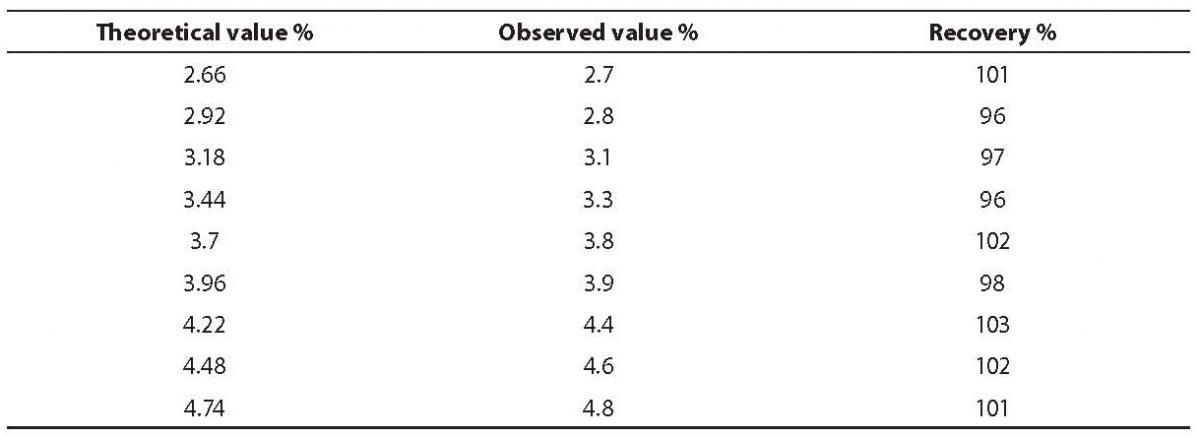
Recovery study by sample mixing was performed to evaluate the response of Primus Ultra2 to increasing concentrations of the analyte, i.e. HbA2.
A normal blood (2.4% HbA2) and β-thalassemia carrier sample (5.0% HbA2) were mixed at the ratios of 9:1, 8:2…1:9 and eleven samples were analyzed.
To make comparison between the theoretical expected values and measured ones, a linear regression test was performed (Method Validator 1.15, P. Marquis statistical software, Metz, France). The line of best fit and linear regression obtained for recovery was Y = 1.089x + 0.01; R2 = 0.992. The measured values correlated well with the expected ones, and the average proportion recovered was 99% (Table 3).
Table 3. Recovery study results
Discussion
Although thalassemia is most common in the Mediterranean basin and Far East countries, due to migration of populations there is virtually no country in the world now in which thalassemia does not affect some percentage of the population. Like other chronic diseases, prevention is important in the overall management of the disease, whereas appropriate screening, detection of patients and counseling of couples at risk are the most important procedures to reduce the morbidity and mortality rates of the disease (18).
β-thalassemia can be diagnosed with confidence when elevated HbA2, RBC count over 5 x 1012/L, microcytosis and normal serum ferritin are present. Although HbA2 concentrations obtained should be interpreted together with other parameters such as iron status, erythrocyte indices or family studies, due to the clinical value of HbA2 assay in the screening for β-thalassemia and the serious consequences of an incorrect diagnosis, the results obtained must be of high quality. With this in mind, it is important to note that no analytical goals for HbA2 measurements have been definitely defined. It is, however, well recognized that the components of biological variation have been used to derive analytical quality specifications for the imprecision, bias and total error of clinical laboratory procedures. The within- and between-subject components of variation, expressed in terms of coefficients of variation (CVw and CVg, respectively) are used to calculate the total error (TE) according to the formula:
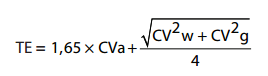
where the goal for imprecision is set on the basis of intra-individual variability CVa < 0.5 CVw (19). Assuming that the intra-individual variation of HbA2 could be similar to that of HbA0 (CVI between 2.8% and 3.4%) (20), the analytical goal for imprecision can be defined between 1.4% and 1.7%. With regard to inter-individual variability, no data are yet available in the literature. A raw estimate could be extrapolated from the reference range in which an average HbA2 can be calculated in normal adults, thus producing a total allowable error of 7.8% (21).
It is important to state that the reference range established in this evaluation (mean 1.9%, range ±2.5 SD 0.9–3.0%), similar to the range provided by the manufacturer (mean 1.9%, range ± 2.5 SD 0.7–3.1%) has lower limits than the range obtained on other HPLC systems (mean 2.5%, range ± 2.5 SD 2.5–3.5%) (22). These values could be explained by the underestimation of the system integrating peak area.
Total allowable error extrapolated from for the reference range 0.9–3.0%, CVg 15.2%, is 6.5%. Total CVs obtained in the imprecision study are lower than this limit.
With respect to HbA2 reproducibility, the Tosoh G7 analyzer was found to yield good within-run imprecision, just a little above the desirable goal for the assay, and a slightly higher between-run imprecision.
The Primus Ultra2 system provides a rapid and reliable separation and determination of the relative percentage of different hemoglobin types, particularly HbA2. The results obtained are accurate and reproducible. There is good differentiation of HbA2values between normal patients and β-thalassemia carriers. The cut off limit can be set at 3.3%, with all patients with values higher than 3.3% being identified as β-thalassemia carriers.
The ease of the operation and the limited technical work make this analyzer suitable for laboratories with a high workload, improving its efficiency and allowing the cost of thalassemia and hemoglobinopathy screening to be reduced.
Our study showed the method to be appropriate for rapid population screening for β-thalassemia carriers, reliable for quantitative determination of HbA2 in blood and suitable for routine clinical laboratory use; however, as in any of the first-level laboratory tests for the characterization of Hb variants, this HPLC system also has potential limits of false interpretation in the presence of an abnormal Hb peak. In these cases, it is recommended that second-level laboratory tests such as molecular analysis and family studies be undertaken in order to achieve definitive identification of the Hb variant.