Introduction
Sulfonylurea stimulates insulin secretion by pancreatic β-cells and is generally used as a first-line treatment for type 2 diabetes. However, after long-term sulfonylurea therapy, some patients do not respond to treatment anymore, which is associated with an increase in blood glucose again, known as secondary sulfonylurea failure (1). Progressive decrease in b-cell mass, deterioration in b-cell secretory reserve, insulin resistance and dysfunction of the proinsulin conversion machinery have been suggested in the pathogenesis of secondary sulfonylurea failure (1,2). The major factors for progressive loss of b-cell function and mass are glucotoxicity, lipotoxicity, proinflammatory cytokines, reactive oxygen species, and accelerated apoptosis (3,4). Impaired b-cell function and possibly b-cell mass appear to be reversible, particularly at early stages of the disease. Short-term intensive insulin therapy is among the interventions to preserve or “rejuvenate” b-cells, and improve insulin sensitivity (2).
Nitric oxide (NO) is a diffusible messenger that has been implicated in numerous physiological and pathological conditions. The interaction of NO with superoxide resulting in the formation of peroxynitrite (ONOO-) and NO inhibition of cytochrome c oxidase causing a decrease in mitochondrial membrane potential and cytochrome c release from mitochondria are proposed mechanisms by which NO induces apoptosis (5-7). An effect of NO on either insulin secretion or resistance is also suggested (8-12).
Adenosine deaminase (ADA; EC 3.5.4.4) catalyzes irreversible hydrolytic deamination of adenosine and deoxyadenosine to yield inosine to deoxyinosine, respectively, as part of purine salvage pathway. It contributes to the regulation of intracellular and extracellular concentrations of adenosine and deoxyadenosine, along with 5’ nucleotidase and adenosine kinase (13). Although the main function of ADA is the development of the immune systems in humans, it also seems to be associated with the differentiation of epithelial cells and monocytes, and inflammation (14). It is increasingly apparent that adenosine is a pleotropic molecule implicated in inflammatory system, apoptosis, cell growth and insulin secretion (14-16).
In this study, we aimed to investigate ADA activity and NO levels as potential mediators of intensive insulin treatment in patients with secondary sulfonylurea failure.
Materials and methods
Subjects
The study was conducted at Gaziantep University, Faculty of Medicine, Department of Internal Medicine, and Department of Biochemistry and Clinical Biochemistry in 2004. Informed consent was obtained from all subjects according to the Helsinki Declaration as revised in 1996. Forty eight consecutive outpatients referred to Endocrinology and Metabolism Clinic in 2004 with a diagnosis of type 2 diabetes mellitus (according to fasting plasma glucose or oral glucose tolerance test as recommended by the American Diabetes Association) and secondary sulfonylurea failure were enrolled in the study. Secondary sulfonylurea failure is defined as insufficient glycemic control (HbA1c≥ 7%) despite receiving maximal dosage of sulfonylureas (240 mg/day gliclazide or 6 mg/day glimepiride) for at least four weeks in patients that have previously achieved sufficient glycemic control with sulfonylureas for a minimum of six months. Exclusion criteria were insulin therapy, irregular use of oral antidiabetics, diabetogenic medications like glucocorticoids, diuretics and b-blockers, conditions associated with medical stress, alcohol intake, pregnancy or lactation, primary sulfonylurea failure, and an additional systemic illness, e.g. renal failure, endocrine disorders, etc. Patients were followed for 24 weeks and 24 were excluded according to the criteria mentioned above. Seventeen female and seven male secondary sulfonylurea failure patients (min-max: 42-73, median: 56, interquartile range: 53-64 years) were recruited. In the study group, diabetes duration ranged from 4 to 32 years. Initially, plasma glucose level was regulated below 8.88 mmol/L by insulin infusion for three days, thereafter maintained with multiple subcutaneous insulin injections for six months: subcutaneous regular insulin injection three times a day plus subcutaneous NPH insulin injection at bed time. During the study period, patients continued their previous diet and exercise programs. Follow-up was performed by outpatient control visits twice in the first month and once in the next five months.
Insulin resistance was assessed using the homeostasis model assessment (HOMA-IR) originally described by Mathew et al. (17). HOMA-IR was calculated using the following formula:
HOMA-IR = fasting glucose (mmol/L) × fasting insulin (mmol/mL)/22.5.
Proteinuria was defined as total protein ≥ 0.15g/24-h urine.
Methods
Blood samples were collected using standard venipuncture technique between 9.30 to 11.00 a.m. after 12-h fast. Serum samples were separated immediately after centrifugation at 4 °C, 2000 g for 10 min and stored at -20 °C until analysis, which was performed in the same run to avoid inter-run analytical variation.
Serum total ADA activity was determined at 37 °C according to the method of Giusti and Galanti based on the Bertholet reaction, i.e. the formation of colored indophenol complexes from ammonia liberated from adenosine, quantified spectrophotometrically (13). Optical density was measured spectrophotometrically at 625 nm in an assay mixture (final volume, 1 mL) containing 12 mM adenosine hemisulfate, 50 mM phosphate buffer (pH 6.5), and 0.05 mL of serum. One unit of ADA is defined as the amount of enzyme required to release 1 μmol/min of ammonia from adenosine at standard assay conditions. Intra-assay and inter-assay precision of the ADA assay was determined from serum pool on 20 replicates in a single run and in 10 different runs yielding CVs of 2.53% and 3.37%, respectively.
Nitric oxide levels were measured with colorimetric assay by the method of Griess reaction as the levels of NO2- and NO3- in serum according to the manufacturer’s instructions (Nitrate/Nitrite colorimetric assay kit, Cayman Chemical Co, Ann Arbor, MI, USA). Urine protein was analyzed with turbidimetric method on a Roche/Hitachi modular P analyzer according to the manufacturer’s instructions (U/CSF Protein, Roche Diagnostics, Mannheim, Germany).
Statistical analysis
Data are presented as median and interquartile range and mean ± SD. Kolmogorov-Smirnov test was applied to test normality of the distributions. Comparison of repeated measures for non-parametric data (NO, ADA, fasting insulin, HOMA-IR, C-peptide, HbA1c, total cholesterol, LDL-c, triglycerides) was performed with Friedman test and post hoc testing with Wilcoxon Signed Rank Test. Comparison of repeated measures for parametric data (FBG) was performed with one-way ANOVA and post hoc testing with Tukey’s post hoc test. Two-tailed P < 0.05 was considered significant. Analyses and illustrations were performed with the SPSS 9.0 (SPSS Inc., Chicago, IL, USA) statistical software.
Results
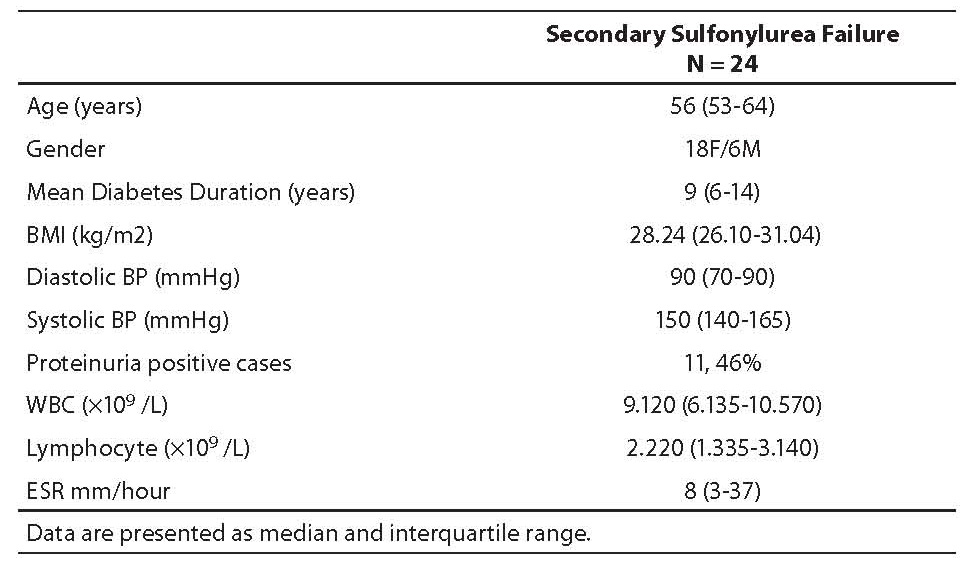
Baseline characteristics of the study subjects, i.e. age, sex, body mass index (BMI), mean diabetes duration, blood pressure (BP), presence of microproteinuria, erythrocyte sedimentation rate (ESR), white blood cell (WBC) and lymphocyte count are presented in Table 1.
Table 1. Baseline characteristics of study subjects (median; 25th-75th percentile)
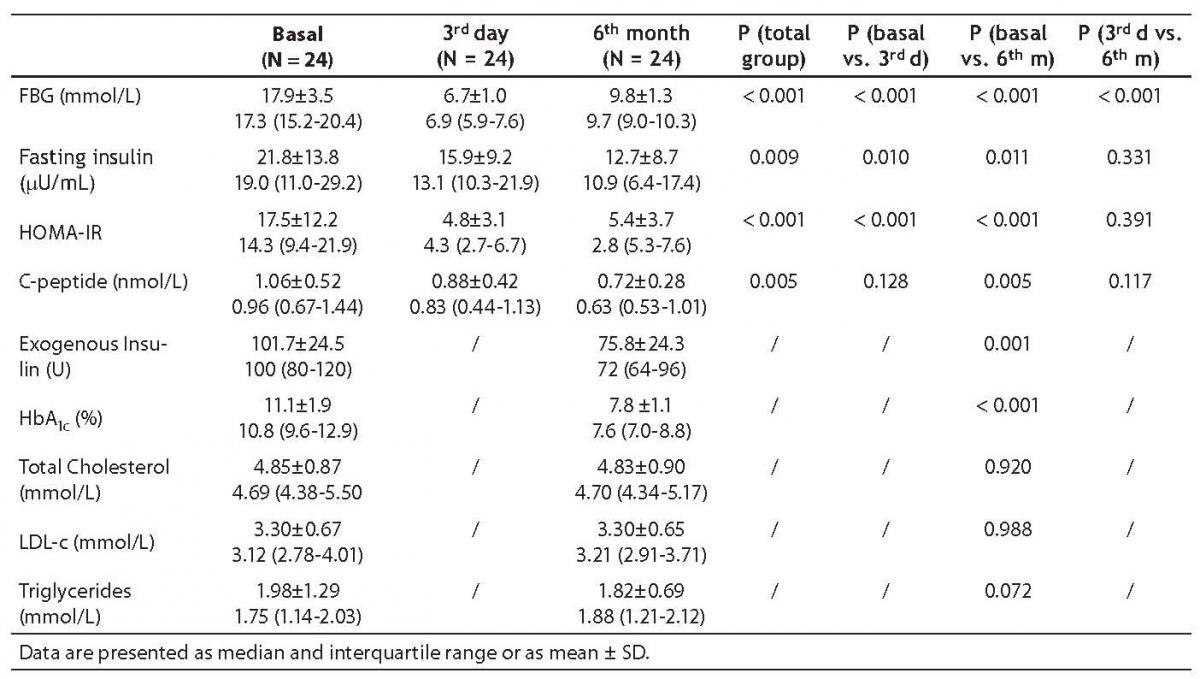
Metabolic changes about diabetes control are summarized in Table 2. Fasting blood glucose (FBG) gradually and significantly decreased during intensive insulin therapy. On day 3 and at 6 months, fasting insulin levels and HOMA-IR decreased as compared to baseline values. The decrease in C-peptide levels became obvious at 6 months. HbA1c and exogenous insulin dose needed to achieve good control significantly decreased. There was no statistically significant difference in total cholesterol and LDL-c. The decrease in plasma triglycerides did not reach statistical significance.
Table 2. Metabolic parameters of diabetes control
In secondary sulfonylurea failure, baseline ADA activity (17.0 [14.6-21.7] U/L) was significantly lower than ADA activity measured on day 3 (20.5 [16.2-23.4] U/L; P = 0.018) and at 6 months (21.2 [16.6-22.5] U/L; P = 0.010) (Table 3, Figures 1 and 2).
There was no statistically significant difference between NO levels determined before (18.8; [11.6-28.4] µmol/L), on day 3 (17.8 [9.7-33.6] µmol/L; P = 0.966) and at 6 months of intensive insulin therapy (21.7 [16.0-33.9] µmol/L; P = 0.230) (Table 3, Figures 3 and 4).
Table 3. Adenosine deaminase (ADA) activity and nitric oxide (NO) level
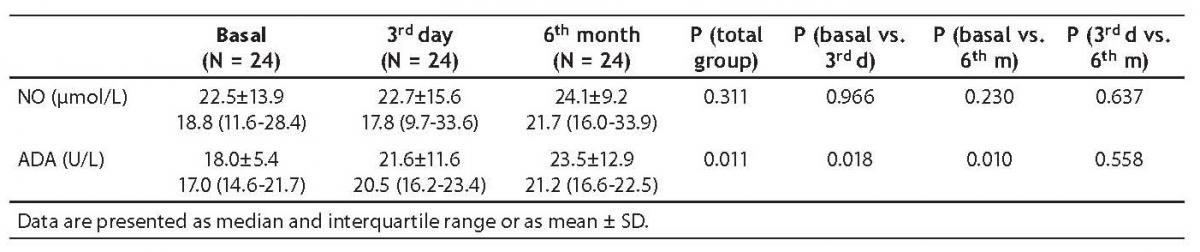
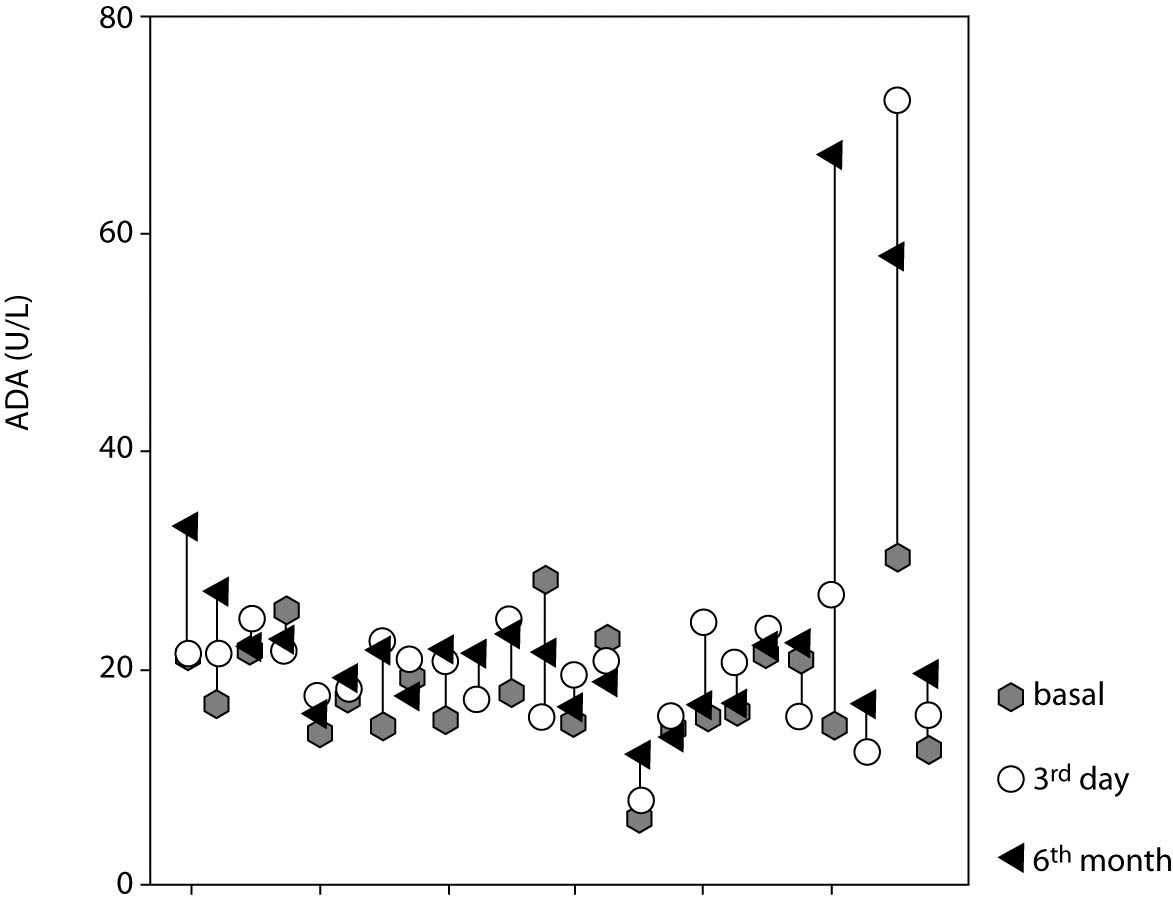
Figure 1. Adenosine deaminase (ADA) activity in individual cases at three different time points.
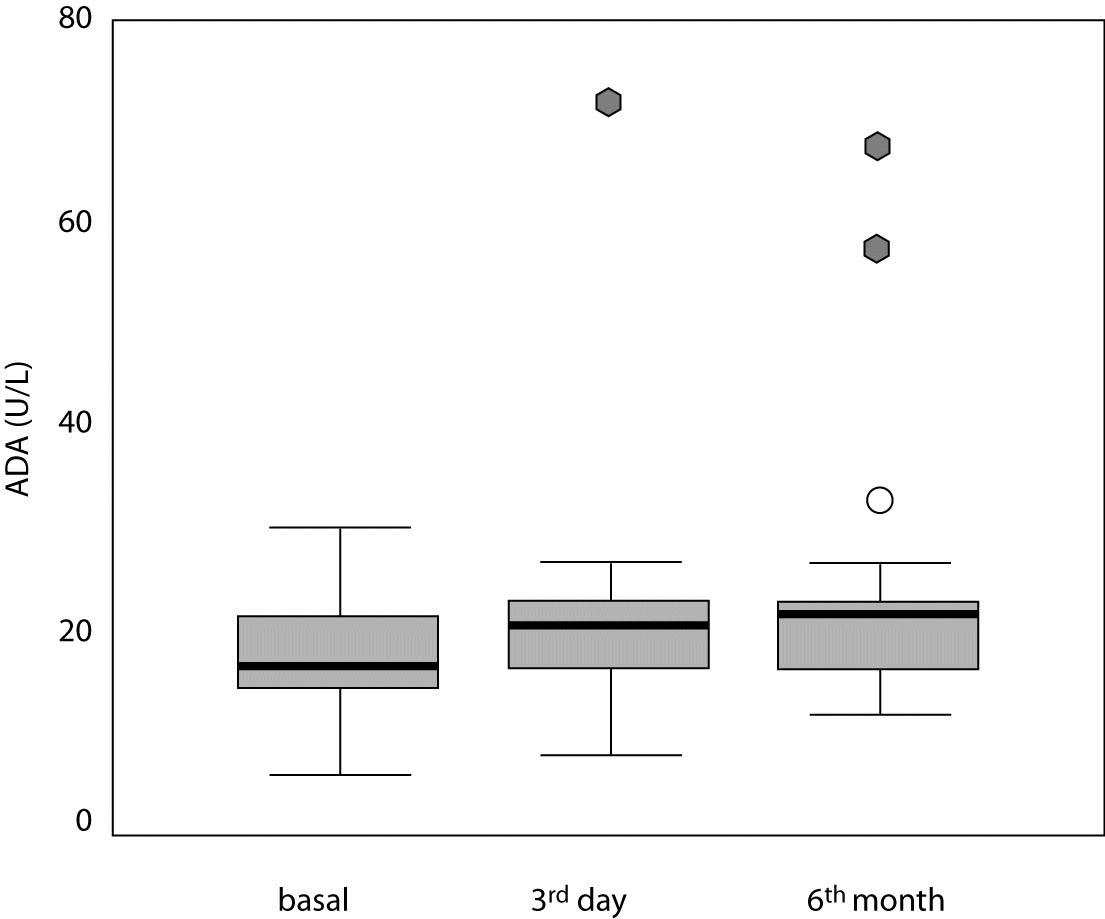
Figure 2. Serum adenosine deaminase (ADA) activity in study patients at three different time points. Central box represents the values from the lower to the upper quartile (25th to 75th percentile). Middle line represents the median. The line extends from the minimum to the maximum value excluding extreme cases and outliers that are displayed as separate points. An outlier is defined as a value between 1.5 and 3 box lengths and an extreme value is defined as more than 3 box lengths from the upper or lower edge of the box. The box length is the interquartile range.
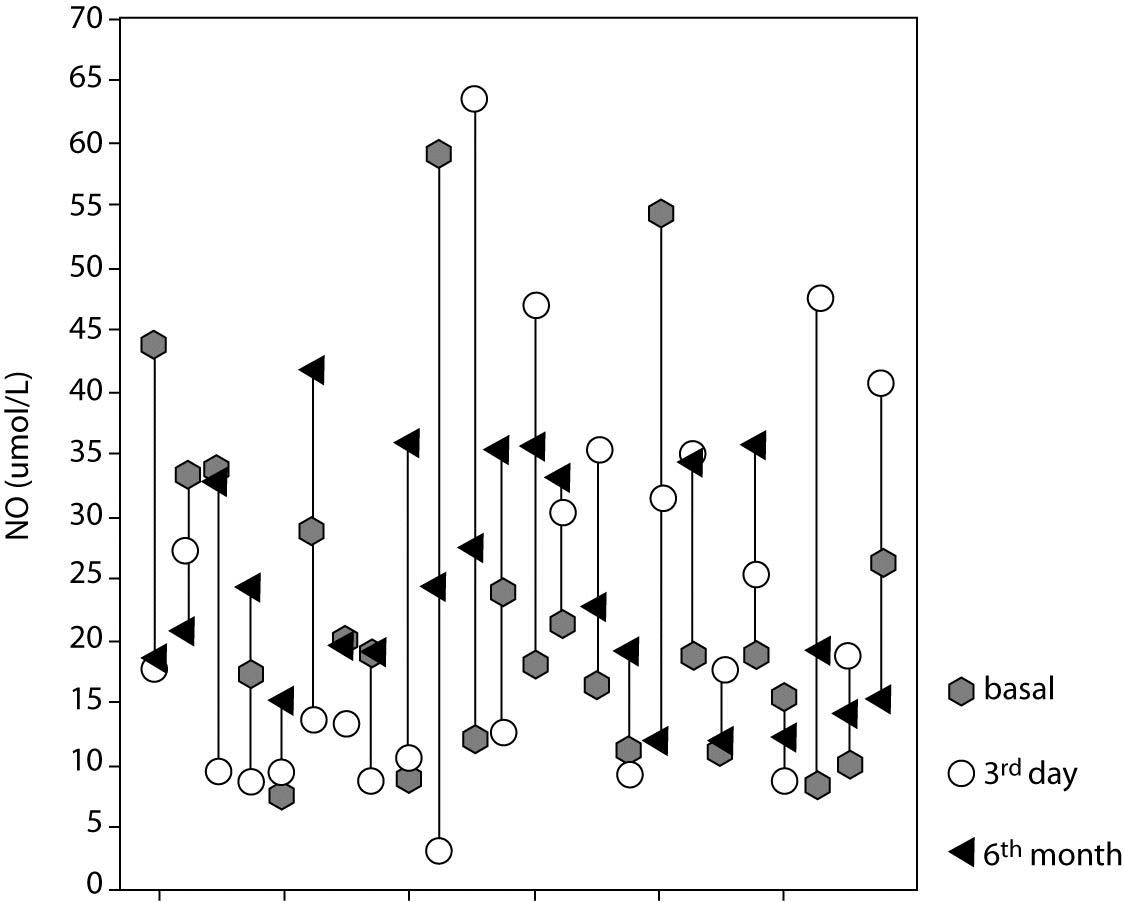
Figure 3. Nitric oxide (NO) level in individual cases at three different time points.
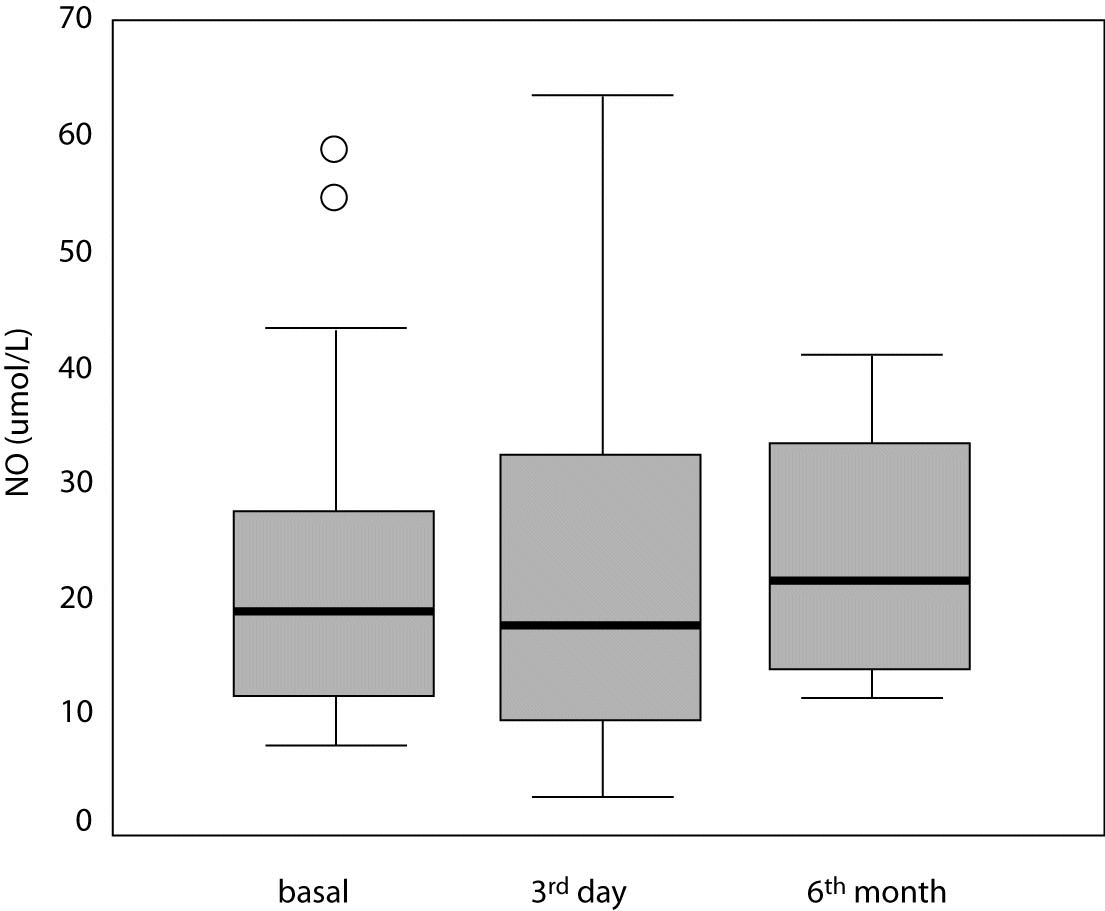
Figure 4. Serum nitric oxide (NO) level in study patients at three different time points. Central box represents the values from the lower to the upper quartile (25th to 75th percentile). Middle line represents the median. The line extends from the minimum to the maximum value excluding extreme cases and outliers that are displayed as separate points. An outlier is defined as a value between 1.5 and 3 box lengths and an extreme value is defined as more than 3 box lengths from the upper or lower edge of the box. The box length is the interquartile range.
Discussion
Despite great progress in the treatment of diabetes, late diabetic complications still remain the principal cause of morbidity and mortality in patients with diabetes mellitus. The Diabetes Control and Complications Trial has demonstrated that tight glycemic control and the resulting long term benefits can be achieved by intensified insulin therapy (18). The present analysis confirmed previous data that intensive insulin therapy can improve glycemic control in secondary sulfonylurea failure as supported by the significant decrease in FBG, HbA1c, HOMA-IR, C-peptide levels and insulin doses required to achieve good metabolic control. In previous studies, one conducted in a surgical and the other in a medical intensive care unit, strict control of blood glucose levels with insulin reduced morbidity plus mortality and morbidity, respectively (19,20). Complications such as severe infections and organ failure were reduced. Several potential mechanisms may explain these benefits, i.e. reduction of systemic inflammation, prevention of immune dysfunction, and protection of the endothelium and mitochondrial ultrastructure (20-25). However, an epidemiological study suggested that the use of intensivetherapy as compared with standard therapy increased mortality and did not significantly reduce majorcardiovascular events (26).
ADA contributes to the regulation of intracellular and extracellular concentrations of adenosine and deoxyadenosine, along with 5’ nucleotidase and adenosine kinase, and increased ADA activity is considered to be a sign of decreased adenosine levels (13,27). Adenosine is elaborated at injured and inflamed sites, and has a central role in the regulation of inflammatory responses and in limiting inflammatory tissue destruction (14). Adenosine modulates the proliferation, survival and apoptosis of many different cell types, ranging from epithelial, endothelial and smooth muscle cells, to the cells of the immune and neural lineages (15). Subsequently, evidence was obtained for extracellular adenosine protective actions in cellular and organ systems, including the brain, kidney, skeletal muscle and adipose tissue. According to our study, ADA activity is increased both in the early and late periods of intensive insulin therapy in secondary sulfonylurea failure. Based on the previous evidence, we suggest that increased ADA activity in this study may have represented disappearance of the initiating medical stress, i.e. oxidative stress, local inflammation, or cell death. It is hypothesized that adenosine is released in response to a wide range of injurious stimuli and mediates an autoregulatory loop, the function of which is to protect organs (14). The term ‘retaliatory metabolite’ was coined by Newby to describe the protective function of adenosine (28).
Literature data on the effects of purinergic agonists on insulin release are controversial. In an experimental study mainly using INS-1 cells, but also using rat pancreatic islets, adenosine inhibited insulin release in a concentration-dependent manner. The inhibitory effect of high ATP concentrations was attributed to its degradation product, namely adenosine because ADA (1 U/mL) abolished the inhibitory effect, indicating involvement of adenosine (27). Furthermore, it was shown that adenosine could act on the endocrine pancreas alpha cell surface to increase glucagon secretion (16). An effect of adenosine on peripheral glucose metabolism has also been suggested. Adenosine increases glycogenolysis and glucose release from the liver (29-31). Therefore, we suggest that increased ADA activity recorded in this study and the accompanying decrease in adenosine concentration may contribute to the benefits of intensive insulin therapy by relieving beta cells’ insulin secretion capacity, amelioration of glucagon secretion and affecting peripheral glucose metabolism.
Nitric oxide affects insulin release, insulin resistance, has proinflammatory, apoptotic and free radical effects through signal transduction and direct posttranslational modifications of proteins, reacting with reactive oxygen species, and interacting with proteins that contain a heme moiety (5,32). Nakada et al. have provided evidence for a concentration-dependent dual effect of NO, i.e. a stimulatory effect at low concentrations and an inhibitory one at high concentrations, on insulin secretion (8). It has been proposed that NO mediates exercise-stimulated glucose transport in skeletal muscle (9,10). Exogenously administered NO, which is generated from the NO donor, sodium nitroprusside, stimulates glucose transport in isolated skeletal muscles by increasing GLUT4 levels at the cell surface (9-11). Part of the mechanism by which insulin increases glucose transport in vivo involves enhanced blood flow and glucose delivery to the muscle, a process mediated by the release of NO from the endothelium (9,12). In this study, systemic NO levels were investigated for its potential role in secondary sulfonylurea failure. However, our study showed that good glycemic control achieved by intensive insulin therapy in secondary sulfonylurea failure had no effect on systemic NO levels. Therefore, the role of NO in amelioration of b-cell function is not supported by the data on systemic NO levels.
The present study had some methodological limitations that may have contributed to the fact that we did not find an impact of intensive insulin therapy on NO levels. We measured NO levels in plasma samples; further studies comprising the mechanisms at the tissue level may provide an insight into the exact role of NO in the setting of secondary sulfonylurea failure. Another potential limitation was the small number of the study subjects. A larger study population and clinical assessment are needed to elucidate whether these results are consistent and have clinical relevance. Also, measurement of adenosine levels may be informative.
In summary, the present study demonstrated that ADA activity is increased both in the early and late periods of intensive insulin therapy in patients with secondary sulfonylurea failure. The implication of NO on intensive insulin therapy in secondary sulfonylurea failure was not verified in this study.