Introduction
The existence of cell free DNA in the human circulatory system has been known since the 1948, when Mandel and Matais managed to prove the existence of nucleic acids in human plasma (1). This great discovery did not attract much attention at that time. A paper from 1977, which suggested the potential application of the cell free DNA in tumor diagnosis and in monitoring the antitumor therapy success rate (2), attracted a somewhat bigger attention of the scientific audience. In 1997, Dennis Lo et al. attracted the majority of interest for cell free DNA research with their discovery of the free fetal DNA presence in maternal blood which opened the possibility for free DNA analysis application in non-invasive prenatal diagnosis (3).
The term ‘’free DNA’’ refers to the compound of DNA fragments detectable in various body fluids. Plasma or serum are most frequently used for that purpose, however, the presence of the free DNA was detected in urine (4-7), saliva (8,9), feces (10), synovial liquid (11), cerebrospinal fluid (12) and peritoneal fluid (13). Normal concentration of the free DNA in healthy individuals varies from 0 to 100 ng/mL of blood, on average 30 ng/mL (14). Majority of the free plasma DNA is double-stranded and consists of DNA molecules sized 0.18-21 kilobase (15). Even though the origin of the free circulating DNA has been researched for the last 30 years, the exact mechanism of its emergence remains unknown. Most researchers agree that it enters in the circulation when a cell dies, whether by necrosis or apoptosis (15,16), however, there is also an opinion that apoptosis and necrosis contribute to the emergence of the free DNA only to a lesser extent while it predominantly occurs as a consequence of spontaneous release of DNA from the living cells (17,18). In the context of free tumor DNA, some opinions state that in addition to the tumor cells themselves, normal cells which are surrounding the tumor also contribute to the level of free DNA (15).
The clearance of such molecules has not yet been clarified. Experimental studies on animal models show that liver, and to a lesser extent kidney, are the organs responsible for the elimination of free DNA from the organism (19). Clearance analysis of free fetal DNA from maternal blood after birth (20) shows that free DNA rapidly clears from the organism (t½=16.3 minutes). Second to blood, the most extensively analyzed source of the free DNA is urine. Experiments performed in mice and humans have shown that the kidney barrier in rodents and humans is permeable for DNA molecules large enough to be analyzed by standard genetic methods (7). This has been additionally proven by the urinary DNA examination (6) which showed two different DNA fractions in urine: the first fraction consists of DNA molecules larger than one kilobase which mainly originate from the cells found in the urinary tract while the second fraction consists of molecules sized 150-250 bp (found in urine supernatant after centrifugation) which at least partially enters into the urine from circulation. This has opened new possibilities for free DNA analysis since the analysis of free urinary DNA has several advantages over free plasma DNA analysis: non-invasive sampling, urine as a sample is less infectious, easier access to a large sample quantity, extraction of DNA from urine is analytically less demanding due to the significantly smaller concentration of proteins.
Healthy people have a very low level of free DNA in circulatory system because dead cells are effectively removed from the organism by phagocytosis. An increased level of free DNA in the circulatory system shows either its increased release from cells or its decreased removal efficiency from the organism or both.
Autoimmune diseases
For many years, free DNA research has been focused on examining the level of free DNA in autoimmune diseases like rheumatoid arthritis (2,11), systemic lupus erythematosus (21-24), systemic sclerosis (25) and primary Sjögren’s syndrome (26).
It has been known for some time that DNA structures that are targeted by auto-antibodies play a central role in systemic lupus erythematosus (SLE) and that DNA-antibody complexes in the circulation are one of the hallmarks of SLE. Investigating whether and to what degree fluctuations in free plasma DNA levels in patients with SLE might correspond to disease severity was the goal of investigations over the past 50 years (21-24). The most recent data seem to exclude measuring free plasma DNA as an inexpensive, simple and quick tool to assess disease activity in patients with SLE (24,25). Further studies on a larger patient population are still needed to confirm these results.
In case of rheumatoid arthritis, Leon et al. (11) have discovered higher concentrations of free DNA in both plasma and synovial liquid than in healthy subjects while the increasing intensity correlated with the symptom severity and the level of the tissue damage. Unlike them, Mosca et al. (26) did not establish the significant difference in free DNA concentration in patients with systemic sclerosis (SS) and in healthy subjects, but based on the free DNA level they could make a difference between patients with active disease and those with the inactive one.
Galeazzi et al. (27) have analyzed both circulating free DNA and DNA extracted from nucleated blood cells in systematic sclerosis and in lupus patients and, by using gel electrophoresis, were able to define the pattern of the DNA, instead of simply dosing its amount in the circulation. They found that SLE and SS have anomalous patterns of DNA both in serum and in the buffy-coat and that these patterns are typical for each disorder. It is possible that understanding the biological significance of the diversity in DNA pattern exhibition in white blood cells may give new insights into the pathophysiology of autoimmune disorders. It is also conceivable that circulating and immune-competent cellular DNA markers might offer the promise of precise quantitative analysis useful for diagnostic purposes, without the need to establish difficult cutoffs as is necessary for protein markers (28).
Tumors
As mentioned earlier here, the first significant step forward in researching free DNA was made with the discovery of increased serum DNA levels in patients suffering from different types of tumors, and these levels were especially high in patients with metastatic tumors. A notable decrease of serum DNA levels was observed following radiotherapy (2). Since increased free DNA levels are not only specific for neoplasms and can also be found in various inflammatory conditions (liver cirrhosis, hepatitis, systemic lupus erythematosus, rheumatoid arthritis), they cannot be considered relevant marker of neoplastic process. In 1989, in a tumor patient’s plasma, DNA with neoplastic characteristics was found (29). Few years later, two papers from 1994 reported that oncogene mutations (in K-ras and N-ras genes) were discovered in the plasma and serum of patients with pancreatic cancer, myelodisplastic syndrome and acute myeloid leukemia (30,31). The latter studies (29,31), which confirmed presence of free DNA derived from tumor cells in plasma/serum of cancer patients, opened up possibilities for detection of more specific changes regarding the tumor process.
The percentage of free DNA of tumor origin is extremely variable. According to some authors, its amount is 10-90% of total free DNA, whereby the highest rates are found in samples with low total DNA levels (15). In other studies, free tumor DNA share in total free DNA amounted to 0.2-10% (29) or 1.9-27% (30).
It is known that tumor cell growth includes genetic and epigenetic changes such as point mutations, chromosomal rearrangements, microsatellite instabilities, promoter region hypermethylation, mitochondrial DNA mutations, and also the presence of some viral DNAs in connection with tumor occurrence (HBV, EBV, HCV, HPV). The said genetic and epigenetic changes, free DNA concentration changes or viral DNA presence have been proven in the last 20 years in various types of cancers: pancreatic (32,33), colonic and rectal (10,34-37), liver (38,39), lung (40,41), head and neck (8,9,42,43), nasopharynx (44), ovaries (13), breast (45,46), prostate (47,48), testicular (49), leukemia (31,50), lymphoma (51,52) etc. The majority of the listed studies point to increased free DNA levels in cancer patients compared to healthy subjects and leaves open the possibility of using free DNA level as an early tumor marker. Hopes are built on the possibility of detecting tumor-specific genome changes, particularly if such detection could be possible before conventional tumor diagnostics. Examples of some tumor-specific genetic and epigenetic changes that were detected in patient’s plasma/serum are presented: N-ras mutations (myelodysplastic syndrome, acute myelogenous leukaemia (31)), K-ras mutations (colorectal carcinoma (37), pancreatic carcinoma (32)), p53 mutations (breast cancer (45), colorectal carcinoma (34)), mutant adenomatous polyposis coli (APC) DNA molecules (colorectal carcinoma (35)), aberrant p16 methylation (hepatocellular carcinoma (38,39), lymphoma (51)), microsatellite alterations (head, neck and lung cancer patients (41,42)), EBV DNA (nasopharyngeal carcinoma (44)), HPV DNA (head and neck squamous cell carcinoma (43)).
Free DNA level changes and the manifestation of tumor-specific genome changes could be useful as treatment success and relapse markers and for discriminating responders from non-responders. For instance, detecting genetic and epigenetic changes in a tumor tissue sample following surgical intervention and regular examining for the presence of such changes in the circulation could enable a very early detection of relapse.
The early detection of cancers through analysis of circulating DNA could have a substantial impact on morbidity and mortality. To achieve this goal, it is essential to determine the number of mutant molecules present in the circulation of cancer patients and to develop methods that are sufficiently sensitive to detect these mutations.
Transplantation
Microchimerism is the presence of a small quantity of foreign cells or foreign DNA in either a tissue or circulation of a person. Naturally acquired microchimerism occurs in a two-way transplancental trafic between the mother and fetus while iatrogenic microchimerism occurs as a consequence of a transfusion or transplantation. Free DNA microchimerism in plasma and urine has been described over 10 years ago in female patients with a transplanted organ from a male donor (4,53).
Kidney transplantation is the most desirable and cost-effective modality of renal-replacement therapy for patients with irreversible chronic kidney failure. Medical causes of early graft dysfunction include a broad spectrum of diagnoses, such as acute rejection, acute tubular necrosis, drug nephrotoxicity and posttransplantation infections. An early and adequate diagnosis is very important because the treatments for some of these pathologies are very different and a mistaken therapeutic management can cause an obvious risk to the recipient. Graft loss caused by acute rejection may be substantially reduced with better control of the adverse immunologic events that occur during the early posttransplantation period (5).
In a study conducted in 2002, urine of female recipients with a male graft was examined for the presence of Y-chromosome (SRY and DYZ-1) while in recipients receiving a HLA mismatched graft the HLA-DR gene (DRB1) was examined (54). Results of the study showed that presence of donor-specific DNA in the urine of the recipient is strongly associated with acute rejection and analysis of DNA derived from donor cells in urine might be an effective and accurate method for the diagnosis of acute rejection of a renal transplant. In all patients with drug-induced renal dysfunction donor-derived DNA was negative (54).
In a similar study from 2009, authors investigated plasma and urine samples from 100 renal transplant recipients obtained during the first 3 months after transplantation. Total free DNA and donor derived free DNA were analyzed by quantitative PCR for the HBB (hemoglobin, beta) and the DYS14 genes, respectively. Plasma total free DNA concentrations increased markedly during acute rejection episodes, often before clinical diagnosis, and returned to reference values after antirejection treatment. In this study, cut-off value for total free DNA concentration was established with diagnostic sensitivity 89% and specificity 85%. Although similar increases were observed during severe posttransplantation infections, use of the combination of total free DNA and procalcitonin (PCT), significantly improved the diagnostic specificity (to 98%; 95% CI, 92-100%), with 97% of the episodes being correctly classified (5).
The same authors performed another study in order to check whether levels of transrenal DNA (Tr-DNA) are influenced by the presence of urinary tract infections (UTI). They found out that when the Tr-DNA quantification is used as a marker for clinical monitoring (i.e. follow up of cancer or transplantation patients), it is important to rule out a concomitant UTI, because a secondary cause of increased concentrations of cell free urine DNA could confound clinical decisions (55).
Quantitative analysis of a total and transplant specific DNA (Tr-DNA) in urine and/or in plasma of the recipient has shown as a good complementary marker of a transplant acceptance or rejection and it can be helpful in the optimizing of immunosuppressive therapy and selective application of a kidney biopsy (5,54,55).
Trauma and sepsis
Knowing the facts that after the severe trauma a systemic anti inflammatory response appears which can lead to the organ failure and that after the increased cell death result is the increase of free DNA level in circulation, Lo et al. have presumed that in patients with a blunt trauma (56) and burns (57) the level of free DNA will be changed. They have determined that free DNA in those patients is increased depending on the severity of injury and can serve as a prognostic marker for the complication development and the recovery length.
A few researchers have studied the changes of free DNA concentration in patients in intensive care units (58-62). They have concluded that the level of free DNA can serve as a prognostic marker for the development of sepsis, organ failure, disease severity and mortality.
Acute myocardial infarction (AIM), acute stroke and acute mesenteric ischemia (AMI)
Elevated level of the cell-free DNA in circulation is connected to the cell death whether as a result of a tissue injury or as a result of an inflammatory reaction. Since the acute myocardial infarction is characterized by a combination of necrosis and apoptosis of the myocytes, several groups of researchers assumed correctly that in patients with AIM level of free DNA in circulation will be elevated (63-66). Results of the mentioned studies showed that level of free DNA in patients with AIM is significantly elevated compared to the healthy subjects and that it has a potentially diagnostic value because patients who have developed complications had higher levels than those with uncomplicated clinical disease course.
Unlike AIM for which there already exist a few very good and routinely used biochemical markers, in the case of acute stroke such markers do not exist in clinical practice. Considering that patophysiology of a stroke includes both cell death and blood-brain barrier dysfunction, a group of researchers from Hongkong (67,68) decided to explore the level of free DNA in plasma of patients with acute stroke. Samples have been collected in a period of 24 hours from the symptom occurrence and results have shown that plasma DNA concentration correlates with the strokes severity and that it can serve as a mortality and morbidity predictor even in those patients with no visible changes detected by neuroimaging techniques.
A group from Spain investigated free plasma DNA levels in order to diagnose patients with acute mesenteric ischemia (AMI) since diagnosing AMI in emergency ward is challenging and quite often results with mortality. They found that plasma DNA levels may be a useful biomarker in predicting the outcome of patients with AMI (69).
Acute pancreatitis (AP)
Only two research groups have investigated level of free DNA in circulation in patients with the acute pancreatitis. In the paper from 2006, authors have shown that the level of free plasma DNA (on the day of admittance and on the fifth day after the admittance) is lower in patients with severe acute pancreatitis compared to control subjects and that it is not possible to differ, by the plasma DNA level, mild from severe illness form (70). In a study published in 2009, authors have shown that the free serum DNA level on the first and fourth day after hospitalization is significantly elevated in those patients who will develop severe form of pancreatitis and that free serum DNA correlates with the pancreatic necrosis level (71). The reason for this disagreement could be different study design (number of participants, number and time of samples per subject, participant’s clinical status) and difference in tissue examined (plasma vs. serum). The most recent study from the second group (72) demonstrate that cell-free DNA measured in both plasma and serum of patients with AP on the first day after hospital admission is significantly higher in patients who developed severe pancreatitis than in those with mild disease. Both plasma and serum levels of free DNA predict severity of AP better than other common markers and scoring systems, but another great value of this new marker is that it predicts disease severity on the first day after hospitalization and enables timely admission to Intensive Care Units (72).
Non-invasive prenatal diagnosis
Notwithstanding their routine application for the past 30 years, modern invasive prenatal diagnostic techniques (such as amniocentesis and chorionic villus sampling) carry a certain risk for the mother and the fetus. The discovery of fetal DNA in maternal blood plasma and serum opened up possibilities for development of non-invasive prenatal diagnostic methods (3). Further breakthroughs revealed its benefits, such as rapid clearance after delivery (20), detectability from 5th week of gestation (73) and easy accessibility (compared to invasive sampling), but also some disadvantages: practically inseparable from maternal free DNA, percentage of free fetal DNA in total free DNA amounts to only 5-10%, depending on week of gestation (74). It originates from syncytiotrophoblast (75) and it was proven to be shorter, more degraded and more hypomethylated compared to maternal free DNA. The presence of free fetal DNA was proven also in other maternal bodily fluids such as amniotic fluid, urine, liquor and peritoneal fluid (74).
Considering that, at the moment, it is technically impossible to separate maternal from fetal free DNA and considering that maternal free DNA interferes with fetal DNA analysis, robust detection and quantification have been achieved when the fetal DNA sequence of interest does not have a maternal counterpart (e.g., Y chromosomal DNA, RhD gene when the mother is RhD negative) by techniques such as real-time polymerase chain reaction (PCR) (76,77).
The first application of free circulating fetal DNA was for the purpose of non-invasive detection of fetal sex. The commonly used markers are SRY, amelogenin, DYS14; the analysis is reliable from 7th week of gestation, and is applied in cases where X-linked diseases is suspected to avoid amniocentesis in case of female fetus, in case of ultrasound-detected indistinguishable or ambiguous sex organs to determine genetic sex and in case of congenital adrenal hyperplasia so that if fetus is a male, dexamethasone therapy can be avoided (76). This analysis is routinely applied in several European countries. In United Kigdom, for example, significant decrease (45%) in a number of invasive tests performed is noted as a consequence of implementation of early noninvasive prenatal fetal sex diagnosis (77).
Other than for the purpose of fetal sex determination, free circulating fetal DNA is typically used for determining fetal RhD status in RhD negative mothers (78-80) to avoid non-selective antenatal immunoprophylaxis.
Measuring free fetal DNA concentration in maternal plasma is the third most common use of free fetal plasma DNA. It is done by measuring quantity of DNA sequences from Y chromosome. Namely, it has been established that its concentration in various forms of pathological pregnancies (preterm labour (81), fetal aneuploidy (82), idiopathic polyhydramnios (83), fetal growth restriction (84), preeclampsia (85), fetal-maternal hemorrhage (86), invasive placenta (87)) is increased so it is investigated as a potential pregnancy pathology marker.
Recently, new universal marker for fetal DNA is discovered and is readily detectable in maternal plasma: hypermethylated RASSF1A. When applied to prenatal RhD genotyping, this marker allows the detection of false-negative results caused by low fetal DNA concentrations in maternal plasma. Hypermethylated RASSF1A marker can also be applied to many other prenatal diagnostic and monitoring scenarios (88).
Since detection of subtle fetal mutations is difficult due to the overwhelming maternal DNA background, possibilities for non-invasive prenatal fetal DNA analysis for various monogenic diseases are limited to paternally inherited or de novo mutations. Until today a few cases of prenatal non-invasive diagnosis of monogenic diseases have been published: ß-thalassemia major (89), myotonic dystrophies (90), achondroplasia (91), Huntington disease (92) and cystic fibrosis (93).
One of the newest applications of free fetal DNA in diagnostics is described in a recently published study. This study demonstrates the first validated non-invasive cell-free fetal DNA fingerprinting method which predicts both gender and which embryo implanted at 9 weeks gestation following multiple embryo transfer (94).
The discovery of the presence of tumor and donor transrenal DNA focused fetal DNA research in the direction of fetal DNA detection in maternal urine, but the study results were contradictory for a long period of time (95). It was not until a recent study, where the size of amplified regions was significantly reduced (96) that the sensitivity of fetal DNA detection in maternal urine improved.
Since fetal DNA concentration is extremely low, Lo et al. detected it in 1997 by using the nested PCR method. With the occurrence of real time PCR instruments, the sensitivity, rapidity and robustness of fetal DNA detection increased.
More recently, new attempts to incorporate mass spectrometric techniques to develop accurate and highly sensitive high-throughput clinical diagnostic tests have been reported (97).
Present fetal DNA studies are based on three basic experimental approaches: enriching free fetal DNA fraction in plasma (98); detecting fetal-specific epigenetic markers or placenta-specific mRNA markers (99-101);developing precise digital PCR (102,103) and second-generation genetic sequencing (104-109).
Other possible applications
Research conducted in 1993 set the level of the plasma DNA as in vivo marker of the cell death in patients older than 68 years of age (110). The highest DNA level was determined in a group of senior subjects with some acute or chronic disease (among them a rate of survival after one month was lower), lower in a group of senior subjects with good health and the lowest level of free DNA was determined in a control group constituted of middle aged subjects.
Level of free plasma DNA was also examined in female patients with endometriosis (111); significantly higher level was determined within the diseased subjects than within the healthy ones. This result is in concordance with the pathophysiological process behind the endometriosis since it is one of the most common benign gynecological proliferations with inflammatory activation in premenopausal women.
A significantly lower level of free plasma DNA was found in patients with Parkinson disease (112) but it is not determined that it represents a confident marker of the disease progression.
In a recently published paper from India, authors managed to distinguish between ataxia patients (Friedreich’s Ataxia and Spinocerebellar Ataxia Types 2 and 12) and healthy controls using free plasma DNA concentration. They hypothesised that significantly higher concentration of plasma DNA appears to be due to neuronal and muscular degeneration in these patients (113).
A group from Shangai performed quantification of circulating cell-free DNA in the serum of patients with obstructive sleep apnea-hypopnea syndrome (OSAHS). They found that the increasing concentration of serum DNA in patients with OSAHS was positively correlated with disease severity. Acoording to them, serum DNA may become an important parameter for monitoring the severity of OSAHS and effectiveness of therapy (114).
One of the research grups tried to investigate the acute effects of a single bout of high-intensive strength training on the production of cell-free plasma DNA (cf-DNA). These results indicate that, cf-DNA (and oxypurines) might be relevant biomarkers for cellular damage, mechanical, energetic, and/or ischemic stress in context with exercise (115).
Besides the level (concentration) measurement and the presence of the specific free human DNA in circulation it is also possible to detect the free parasitic DNA. That is useful because, in some clinical stages of the disease, it is difficult to detect parasitic infection (schistosomiasis) (116).
Discussion and conclusion
In the past decade, cell free DNA, or circulating cell free DNA, or cell free circulating DNA, isolated from body fluids such as plasma/serum/urine has emerged as an important tool for clinical diagnostics. The molecular biology of circulating cell free DNA is not completely understood but there is currently an increased effort to understand the origin, mechanism of its circulation and sensitive characterization for the development of diagnostic applications. There has been considerable progress towards these goals using real-time polymerase chain reaction technique. More recently, new attempts to incorporate mass spectrometric techniques to develop accurate and highly sensitive high-throughput clinical diagnostic tests have been reported (Table 1).
Aim of this brief overview was to present various clinical fields and pathological states in which analysis of free DNA could be of interest (Table 2). Just by browsing at the quantity of published scientific papers on the topic of free DNA it is easy to conclude that researchers and clinicians show (and share) a great interest for this scientific field. Because of that, one can expect that this analyte will soon became part of the routine clinical care and management of the patient. In order for this to happen, few important issues need to be solved.
The first encountered problem when comparing and evaluating results of various studies is the non-standardization of the methods used. Different groups have used different methods for DNA isolation, quantification and detection, with each of them having its sensitivity and specificity. For example, previously used methods for DNA detection had a significantly lower sensitivity compared to nowadays broadly spread real time PCR. Nowadays, the majority of researchers use commercially available columns for DNA isolation based on the ion exchange process of binding DNA. And even when the same extraction method was used, starting sample volumes and volume of the elution buffers were different.
Second issue is preanalytical: the non-existence of consensus on the preferred tissue for analysis (serum vs. plasma) and on the centrifugation speed during the serum/plasma separation. In the early years of free DNA research, serum was more favorable compared to plasma. Nowadays, plasma DNA analysis is more common and more widespread, mainly because in that way it is possible to avoid false higher free DNA values which are result of cell degradation during the process of clot formation.
Except for the sample type and centrifugation protocol, sample collecting tubes are also important because they can significantly influence free DNA yield (117). When samples can be processed within eight hours of blood draw, K3EDTA tubes can be used. Prolonged transfer times in K3EDTA tubes should be avoided as the proportion of fetal DNA present decreases significantly; in these situations the use of cell stabilising tubes is preferable.
Third problem is the use of different standards for free DNA quantification, commercially available or prepared in house. Consecutively, quantitative analysis results have been expressed in two ways: in ge/mL (ge = genome equivalent) or in ng/mL. Although those units could be mutually converted by use of the conversion factor of one diploid genome being equivalent to 6.6 pg of DNA (118), it additionally complicates the comparison of results from different studies.
One should be aware that data obtained as a result of quantitative analysis of the complete free DNA is extremely nonspecific because elevated free DNA level has been noted in different pathological states (inflammation, autoimmune process, different acute states, malignancy etc.) which sometimes do not exclude one another, especially because not even the mechanism of origin nor free DNA elimination has been completely clarified. Interestingly, only few researchers have considered the influence of liver and kidney insufficiency on the quantity of free DNA, which could have the most significant influence on patients with severe sepsis, trauma or organ failure (patients in Intensive care units). So, application of quantitative DNA analysis in acute states for diagnostic purposes will hardly become part of the routine practice for two main reasons: nespecificity of the results and length of the analysis (2-3 hours). However, this result could serve as an excellent prognostic marker.
In the field of free tumor DNA, its value is rarely correlated with the value of the other tumor markers. In order for analysis of genetic and epigenetic changes to serve in the early tumor detection, it is necessary to be very familiar with the genome changes specific for a certain type and localization of the tumor because those already known are often characteristic for different types of tumors.
According to the latest research, application of DNA analysis in transplant patients has a great potential for utilization in the clinical laboratory. In fact, today’s markers of rejection of a kidney transplant (urea and creatinine) are not particularly sensitive and biopsy, even though a ‘’golden standard’’, is a diagnostic procedure which should be applied selectively. Measurement of free DNA in transplant patients in certain time intervals (with a marker like procalcitonin for differential diagnostic exclusion of patients with urinary infection (55)) would be useful for monitoring the state of the kidney transplant as well as the selective biopsy application.
Although free DNA research is being done for many years, progress in a diagnostic sense is not going according to the plan. One reason for slow utilization of this potential new clinical marker is that majority of the research was done on a smaller number of subjects (20-70), rarely over 200, and so far there were only few large clinical and multicenter studies (106-109). Therefore the power of statistical tests used in the analysis is questionable as well as the conclusions brought on the basis of it.
In addition, reference intervals for the healthy population are still not available. Since the creation of reference intervals requires careful planning, monitoring and documentation of every aspect of the study and characterization of variations attributable to the pre-analytical and analytical factors, only future large studies can provide data for establishing them (119).
Undoubtedly, the greatest progress has been achieved in the field of non invasive prenatal diagnosis (NIPD). While in some other diagnostic fields, analysis of free DNA is done exclusively in research institutions, free DNA analysis for the purpose of non invasive sex determination and fetal RhD status determination is conducted as a routine practice in some EU countries (77,80).
Due to the technical impossibility of separating maternal from fetal DNA, application of free fetal DNA analysis has its limitations: non existence of the positive control for extraction and amplification of fetal DNA, analysis is limited by fetal gender and knowing of the parental genotypes, it is possible to detect only those mutations which are parentally inherited or occurred de novo.
As mentioned earlier, a new test for the detection of fetal DNA in maternal plasma was developed; it is based on the detection of a hypermethylated placental (fetal) DNA sequence in the maternal circulation. In contrast to existing fetal DNA markers that detect Y-chromosomal sequences and genetic variations between the fetus and mother, this marker (RASSF1A promoter) is applicable to all pregnancies irrespective of the sex and genetic variations of the fetus (88).
During the last three years the development of digital PCR and second generation genetic sequencing (103,104), analytical techniques which enable non invasive prenatal diagnosis of aneuploidy, has overcome these limitations. These techniques enable measurement of relative amount of free DNA from chromosome 21 (direct measurement of total number of copies from chromosome 21 according to the referral chromosome) in such a way that it is possible to estimate number of chromosomes 21 (or any other) in a sample that contains fetal DNA (Table 1).
Table 1. Methods used for free DNA analysis.
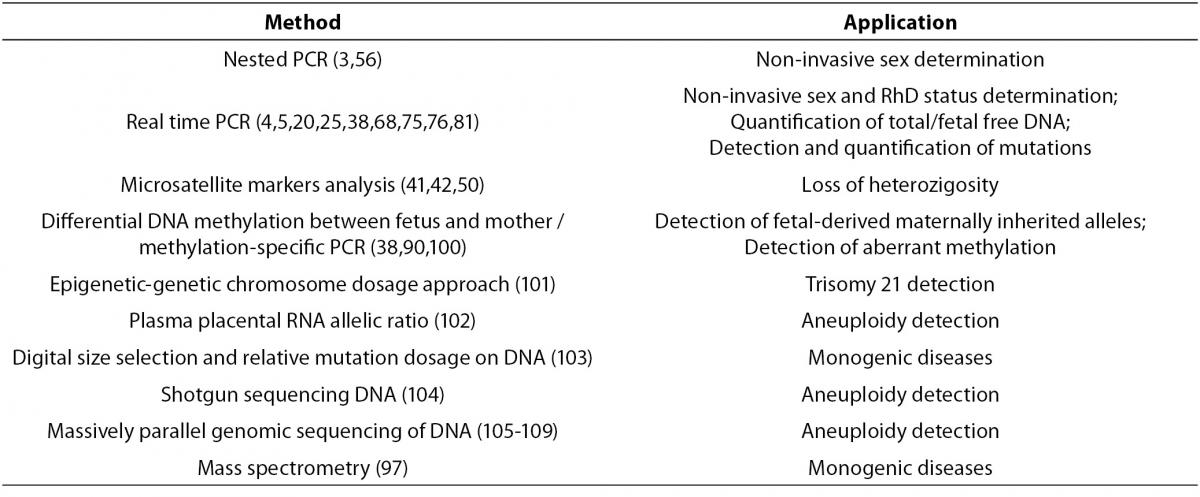
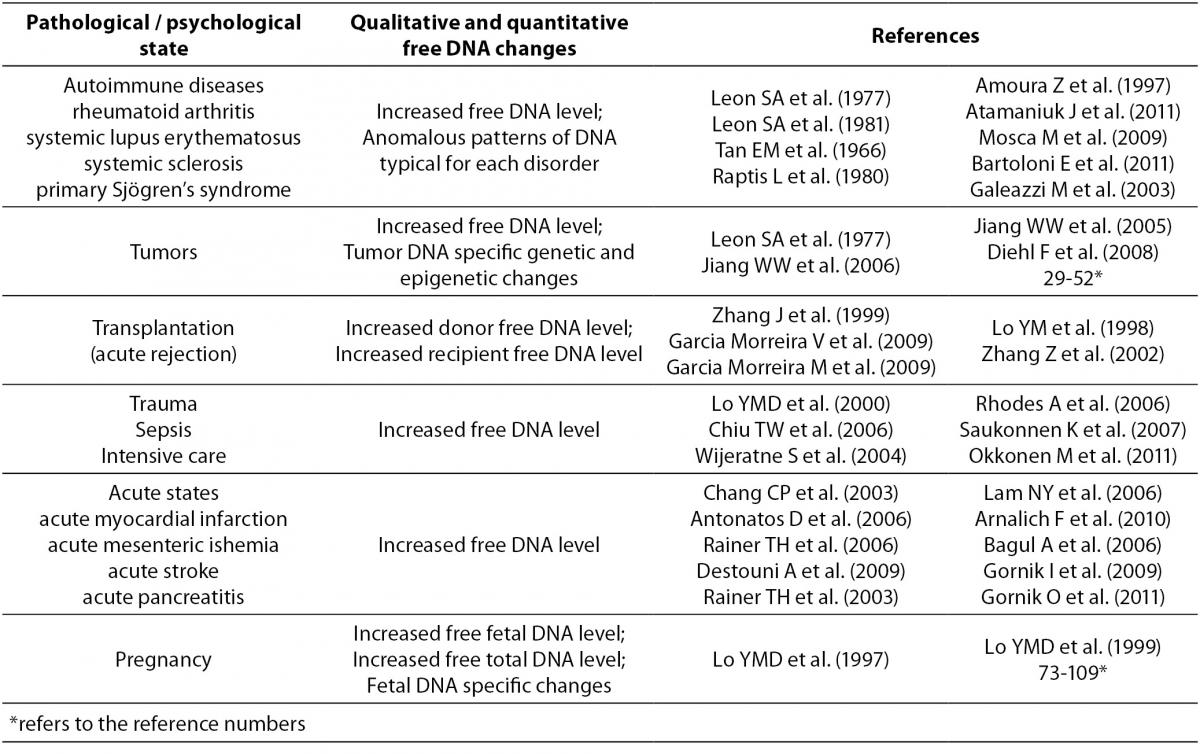
Table 2. Qualitative and quantitative changes of free DNA in different pathological states
The advent of massively parallel sequencing has given us a quantitative and powerful tool for studying circulating DNA on a genome-wide level (120). Using this approach, fetal chromosomal aneuploidies can be robustly detected using maternal plasma. Furthermore, a genome-wide genetic map of a fetus can also be constructed using this approach (120). This method has also allowed one to identify tumor-associated chromosomal translocations, which can then be detected in plasma. Current deficiencies of these methods are their expensiveness, longevity and analysis complexity, however, with the further technological progress all of these disadvantages should be overcomed and non invasive prenatal detection of most common aneuploidy enabled.
As cell-free fetal DNA testing continues to be developed and translated, significant ethical, legal and social questions will arise that will need to be addressed by those with a stake in the use of this technology. Since non-invasive prenatal diagnosis is likely to be introduced in the near future, this will raise important ethical questions concerning meaningful reproductive choice, the autonomy rights of future children, equity of access and the proportionality of testing. It will probably be easier to get cell-free fetal DNA testing for chromosomal abnormalities and single-gene disorders, but caution should be taken with respect to determination of sex and behavioral or late-onset conditions.
Despite all of the earlier denominated issues, free DNA research is continuing in the form of the large scale clinical studies which would at the end evaluate its potential application as new analyte in clinical laboratory diagnostics. My personal favorite is field of non-invasive prenatal diagnosis, where the breakthrough will be the biggest and will lead to abandoning invasive methods in prenatal diagnostics.