Introduction
Uric acid is a weak organic acid, the end product of purine nucleotides degradation (1). Source of purine nucleotides are ingestion, endogenous synthesis of purines from nonpurine precursors, and reutilization of preformed purine compounds (2). Degradation of purine nucleotides (Figure 1) starts with nucleotidase activity in reaction which releases phosphate from nucleotides and produces nucleosides, adenosine and guanosine (3). Adenosine is than deaminated into inosine in reaction catalyzed by adenosine deaminase. Purine nucleotide phosphorilase hydrolyses ribose group from inosine and guanosine to produce xypoxantine and guanine, respectively (3). Guanine is deaminated to xsantine. Xantine oxidoreductase is widely distributed enzyme. It oxidases hypoxantine to xsantine, and finally xantine to uric acid in the liver, gut, lung, kidney, heart, brain and plasma (4).
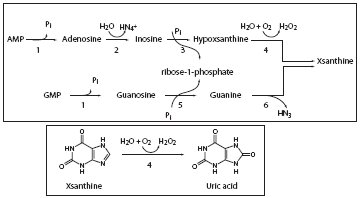
Figure 1. Degradation of purine nucleotides and formation of uric acid. 1 - Nucleotidase; 2 - Adenosine deaminase; 3 - Inosine phosphorilase; 4 - Xsanthine oxidase; 5 - Purine nucleosidase phosphorilase; 6 - Guanine deaminase.
Under the steady-state conditions the production of uric acid is in balance with the uric acid disposal (5). Deposition of monosodium urate monohydrate microcrystals or uric acid in various parts of the body leads to the development of gout, common painful rheumatic disease (6).Gout is not only disorder which might have altered concentrations of serum uric acid (SUA). The enzymes involved in uric acid production are also responsible for oxidative stress (1) and it was evidenced that SUA might be dependently or independently related to different multifactorial disordes. Elevated SUA positively correlates with development and progression of cardiovascular diseases (7). There is an evidence of positive association between SUA and risk of major coronary heart disease events. SUA is significantly positive correlated with hypertension and renal diseases (8). Nephrolithiasis of uric acid origin, as well as hyperuricemia, is more common among patients with the metabolic syndrome (MetS) and obesity (9).There is also a controversial opinion about prooxidative and antioxidant properties of uric acid.Uric acid is a powerful free radical scavenger in humans (10) but it also represents a marker for high levels of damaging oxidative stress associated with increased xanthine oxidase activity (11). Because of all mentioned facts, it is reasonable to determinate SUA concentrations in different conditions.
Determination of uric acid concentration includes phosphotungistic acid methods, uricase methods (in wide usage), high-performance liquid chromatograpy methods (reference methods) and dry chemistry systems (12).
The subject of this review is to present: a) different conditions with observed hyperuricemia; b) correlations and causal connections of uric acid and different disorders like cardiovascular and cerebrovascular diseases, hypertension and metabolic syndrome; c) the protective and pathogenic role of uric acid in those conditions; and d) advantage for widespread determination of uric acid.
Uric acid precipitation and gout
Gout is common painful rheumatic disease, resulting from the deposition of monosodium urate monohydrate microcrystals or uric acid in various parts of the body, primarily in joints and surrounding tissues due to overproduction or underexcretion of poorly soluble uric acid which is the end product of endogenous and dietary purine metabolism in humans (6). Uric acid ionizes with pKa at pH 5.75 (6). In the extracellular fluid, at physiological pH of 7.4, uric acid is mainly in the ionized form of urate, while in the urine, which is usually acidic, the un-ionized uric acid form predominates (13). Risk for crystal formation and precipitation increases when urate concentrations exceed 380 μmol/L. Uric acid concentration is controlled by synthesis and cell turnover and in the other hand by the rate of destruction, excretion and reabsorption by the kidney. In urine, at pH 5.0, predominates undissociated uric acid formed from urinary urate in the renal tubule. In healthy individuals two-thirds of uric acid is excreted by the kidney and one-third by the intestine with breakdown of urate by gut bacteria. Purine ingestion, endogenous synthesis of purines from nonpurine precursors, and reutilization of preformed purine compounds are the sources of urate production, an overall process that under steady-state conditions is in balance with the uric acid disposal (2). Human and higher primates lack peroxisomal enzyme urate oxidase (uricase) (14) that catalyzes degradation of uric acid to more soluble product allantoin that is easily eliminated by renal excretion in the most other mammals because the genes for uricase have mutated and become dysfunctional (15). Consequently, serum urate levels in humans are to its limit of solubility. Hyperuricaemia is a risk factor for gout, but gout can occur where serum uric acid levels are low (16). Also, individual can have uric acid concentrations above normal and not have gout. The prevalence of gout varies with age and gender, and is more common in older individuals (16). Gout is more common in men than in women, but women become increasingly susceptible to gout after menopause because of increased uricemia parallel with decreasing of estrogen levels (17). Risk factors for gout are also obesity, metabolic syndrome, diuretic therapy, high-purine diet and alcohol intake. Gout can be classified as primary or secondary, depending on the presence / absence of an identified cause of hyperuricemia. Primary gout is more common, without identifiable underlying disease causing of the hyperuricemia (18). Secondary gout, which is less common, can be a result of many conditions (6,19). Alcohol intoxication causes lactic acidemia and reduces renal urate excretion (19). Thiazide and loop diuretics (antihypertensives), increase serum urate levels by interfering with renal tubular ion transport (19). Several immunosuppressant like cyclosporine, used in patients undergoing solid organ transplantation, substantially reduces the renal clearance of serum urate (19). Malignant diseases are characterized with intensive purine metabolism which might lead to hyperuricemia and gout. In patient with renal failure, hyperuricemia is, behind the other, one of characteristic caused by impaired kidney function (20). Gout can be the consequence of hereditary interstitial kidney disease, because of renal failure which is caused with this hereditary disorder (21).
SUA concentration might be decreased by usage of a number of pharmacologic agents, dietary and other risk factor modifications. Gout management is very difficult because gout and hyperuricemia may have a great impact in some co morbidities, but on contrary higher urate levels potentially have some beneficial effects (22). Namely, uric acid itself may serve as an immune system stimulant, urate is a potent antioxidant, it helps to maintain blood pressure in a salt-poor environment and it impliessome benefits in several disease of central nervous system, probably because of its antioxidant properties (22).
It is also important to define if hyperuricemia and gout are either a consequence or a cause of these related conditions. The treatment of gout includes the changes in lifestyle, nutrition, and adjunctive therapies as well as different classes of drugs which are approved for lowering urate levels: xanthine oxidase inhibitors, uricosuric agents, uricase agents and some which are still under development, such are inhibitors of urate transporter, purine nucleoside phosphorylase and interleukin-1 antagonists (23). The safety and efficacy of combination therapies for the treatment of gout still remained unclear. Unfortunately, lowering of urate concentration does not relive gout symptoms and urate-lowering efficacy is poor as well as clinical outcomes in gout patients (24). Hyperuricemia with gout, isolated gout without hyperiuricemia or hyperuricemia without gout are conditions which require ordinarily monitoring. Therapy indications, as well as changes in lifestyle, depend also on the other risk factors.
Uric acid and oxidative stress
Oxidative stress is considered to be one of the main reasons of cells function impairment. It is a condition of excessive production of free radicals and reactive oxygen species (ROS), as well as reduced antioxidative system, due to decreased intake of antioxidants or their excessive consumption (25). It is also a condition in which oxido-reductive processes in cells are turned in favor of oxidation due to excessive formation of free radicals and reactive oxygen species (ROS) (25). Uric acid is a product of purine nucleotides catabolism in the process catalyzed by liver enzyme xanthine oxidoreductase (XOR), which enables the oxidation of hypoxanthine to xanthine and its further oxidation to uric acid (26). During the production of uric acid catalyzed by xsanthine-oxidase, ROS are generated as by product,which have a significant role in the increased vascular oxidative stress (26).
XOR is a hepatic enzyme which catalyzes the production of uric acid, nitric oxide, and reactive oxygen species, which potentially damage deoxyribonucleic acid, ribonucleic acid and proteins, inactivate enzymes, oxidize amino acids and convert poly-unsaturated fatty acids to lipids (27). XOR exists in two inter-convertible forms: XO-xantine oxidase and XDH-xanthine dehydrogenase (1,28). Humane XOR exists in vivo as the dehydrogenase form but is easily converted to XO by oxidation of the sulfhydryl residues or by proteolysis (4).There is the difference in substrate affinity of XO and XDH subforms (Table 1). XDH preferentially reduces NAD+, whereas XO cannot reduce NAD+, preferring molecular oxygen (28).Reduction of molecular oxygen by either form of the enzyme yields superoxide and hydrogen peroxide (1,4,28), Uric acid is recognized as a marker of oxidative stress, but also as a protective factor acting as an antioxidant (10,29).
Table 1. Xanthine oxidoreductase (XOR) isoforms and characteristic reactions (4,28).

Regarding XO role in oxidative stress and pathophysiology of CVD, most therapeutic approaches are oriented on inhibiting the XO activity and disabling free radicals accumulation as a feasible target. Some studies showed that therapeutic use of allopurinol, xanthine oxidase inhibitor, which reduces serum levels of uric acid, show protective effects in situations associated with oxidative stress (27,30), because of decreasing of ROS. In clinical studies, for example in coronary bypass surgery, the protective effects of allopurinol are noticed as decreasedhospital mortality rate, increased cardiac index, better postoperative recovery and reduced lipid peroxidation (4). In heart failure allopurinol improves myocardial contractility by restoring myocardial calcium sensitivity and β-adrenergic responsiveness (31). The other explanation of benefitial allopurinol effect considers the effects on vascular endothelial function. Xanthine oxidase plays a key role in mediating intermittent hypoxia-induced vascular dysfunction and administration of allopurinol might prevent it (32). The investigations on animal models in the most cases also confirmed the beneficial effect of allopurinol after induction of myocardial infarction (4) with reducing myocardial damage, better recovery of ventricular function, better preservation of cellular ATP levels and mitochondrial ATP generation during ischemia and prevention of the decrease in left ventricular pressure. The beneficial effects of allopurinol may be unrelated to the inhibition of the XO, and decreasing the free radicals.The effects of allopurinol action as a xanthine-oxidase inhibitor are increasing of intermediates, hypoxanthine and xanthine, and decreasing of final product uric-acid (33). Taking into account that xanthine derivatives are widely used medicaments in treatment of different disease (inflammatory diseases, behavioral, neurodegenerative diseases, renal diseases, respiratory diseases, cancer pain etc, (34) the effect of xanthine or hypoxhantine should be considered in explanations of the mechanisms involved in effects of XOR inhibition (35).It is well known that widely used and examinated xanthine derivative is caffeine. The biological effects of caffeine and other xanthines have many molecular targets. Xanthines are antagonists of adenosine receptors, inhibitors of phosphodiesterases, antagonists of GABA receptors and sensibilators of calcium releaseryanodine-sensitive channels in the sarcoplasmic and endoplasmic reticulum (34). It appeared that caffeine also has stimulatory effects on Mg-ATPase and calcium sensitivity in cardiac myofibrils (36).
Uric acid itself also can have an impact in oxidative stress. Experimental data showed that NO- labeled 1,3-15N2-uric acid (15N2-UA) under anaerobic conditions in several different media, including human plasma and endothelial cell lysates results with production of 5-aminouracil (5-AU) and 6-aminouracil (6-AU) (37). These findings provide a mechanism for how uric acid may inhibit endothelial function by inhibition of NO-function under conditions of oxidative stress (37). Recent clinical studies demonstrated that high-dose allopurinol usage was associated with a lower risk of cardiovascular events and all-cause mortality (38), and that profoundly reduces vascular tissue oxidative stress and improves vascular/endothelial dysfunction (39).
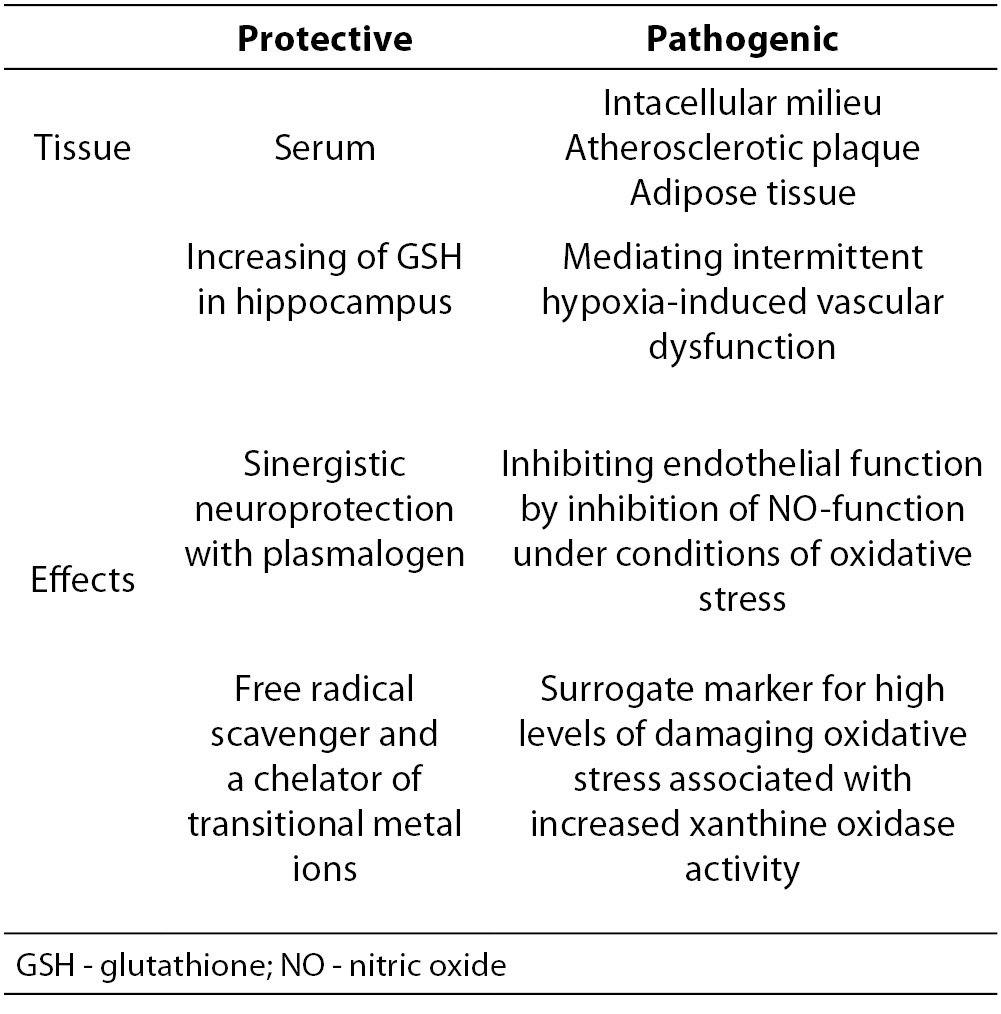
Elevated serum concentration of uric acid are associated with increased risk for cardiovascular disease (CVD) (40), but there are also evidence of their potential role as antioxidants in CVD (41). Some protective and pathogenic roles of uric acid are summarized in table 2. Uric acid acts as an ‘‘antioxidant’’ substance with acting as a free radical scavenger and a chelator of transitional metal ions which are converted to poorly reactive forms (42).Both in vitro and in vivo studies confirmed that uric acid is a powerful free radical scavenger in humans (10). Antioxidant properties of uric acid could be expected to offer a number of benefits within the cardiovascular system or other oxidative stress related disorders. Thus, the role of high uric acid levels (supportive or protective) in development of cardiovascular disease is still unclear, and the explanations lie in different approach of in vitro experimental studies, as well as in different design of clinical studies.
Table 2. Some protective and pathogenic roles of uric acid (1,8,30).
The role of uric acid in cardiovascular and cerebrovascular disorders
Pathophisiology of atherosclerosis and its consequences coronary artery disease, cererebrovascular disease and finally acute myocardial infarction (AMI) and stroke are related to oxidative stress. Interrelationship between concentrations of SUA and cardiovascular or cerebrovascular diseases has been observed for a long time. Most of the cardiovascular risk factors are in potentially overlapping with SUA concentrations (43-45). The positive association of hyperuricemia with obesity, impaired glucose tolerance, hypertension and history of heart disease was observed on a large Finland cohort population (aged 40-69) (43). The study also showed significantly higher total mortality of hyperuricemic men and women in 5 years than in normouricemics (43). Retrospective analysis of general Italian population (N = 10,181) showed that study subjects with hyperuricemia were characterized by significantly higher prevalence of abnormal values of fasting plasma glucose and triglycerides as compared to subjects with reference concentrations (45).
The most important and well evidenced is possible predictive role of uric acid in predicting short-term outcome (mortality) in AMI patients and stroke (43,46).
Different prospective studies exploring the interrelationship between serum uric acid and cardiovascular outcomes in various categories of subjects are summarized by Strazzullo and Puig (30). In general conclusions, in population samples with relatively low risk of CVD, SUA is a very weak predictor of CVD, but it is a significant independent predictor among subjects at high or very high risk (30).Based on the study design it has been shown that hyperuricemia might be independent factor of cardiovascular events (46-48) primarily in subject groups with high cardiovascular risk (angiographically proven CHD,congestive heart failure and stroke survivors) (30). The first large prospective study was established on 83,683 Austrian men cohort who was prospectively followed for a median of 13.6 years (49). This investigation confirmed the independent relationship between elevated SUA and mortality from coronary heart failure and stroke, but the association of SUA with mortality from acute, subacute, or chronic forms of coronary heart disease (CHD) after adjustment for potential confounding factors was absent.
Cross-sectional population-based, follow-up study of epidemiological data (N = 5926 US-subjects, aged 25 to 74) showed that increased serum uric acid levels are independently and significantly associated with risk of cardiovascular mortality (46). In cross-sectional study on 982 Turkish patients,whom coronary angiography was performed for the suspicion of CAD (47), SUA concentrations wereindependently associated with the severity of coronary atherosclerotic plaques. In 212 Spanish patients, who were hospitalized because of acute HF and left ventricular systolic dysfunction, the presence of hyperuricemia was associated with a higher risk of death and/or new hospitalizations in the long-term, independently of ventricular function, renal function and functional ability (50). Results of some other studies, on contrary suggest that serum concentrations of uric acid were not an independent risk factor for CVD. In retrospective study, which included 716 Korean consecutive patients who underwent coronary angiography after adjusting for age, diabetes, smoking, cholesterol, and metabolic syndrome there were no independent association of SUA (51). Any apparent association with these outcomes is probably due to the association of uric acid level with other risk factors (52). This was shown especially in thestudies of general population and studies involving patients with history of arterial hypertension, which didn’t show independent association with CVD (30). Different investigations also indicate that uric acid does not have a causal role in the development of coronary heart disease or death from CVD. The contribution of SUA in atherosclerosis development through effects on arterial calcification was absent in the multicentre American Family Heart Study on population-based cohorts in the US, which was designed to assess risk factors for heart disease, the association between SUA and carotid atherosclerotic plaques (53). It is evidenced that there is no consensus about its action in cardiovascular disease, although the confirmation of active participation of uric acid in heart failure exists (54). However, investigation based on large prospective studies and cross sectional follow-up studies confirmed the independent association of SUA with cardiovascular diseases. On contrary, retrospective studies, studies on general population and those performed on smaller number of study participants did not always provided the similar results.
Beside the study design, different observation results also might be explained on cellular level. Namely, there is a difference between uric acid in serum and intracellular uric acid or highly hydrophobic areas such are atherosclerotic plaquesand adipose tissue. Antioxidant activity is evidenced in serum while it seems that intracellular uric acid contributes in pathophysiology of heart failure (54).
It is evidenced from the literature data that these controversies are very interesting subject for discussion between different scientific groups (55,56). Conflicting conclusions of the different studies are the consequence of differences in the compositions of the population’s studies (number, treatment, biochemical and clinical data), length of follow up, analysis of the different CAD-risk variables and corrections according those variables (49). Proctor et al. reported on the association between high urate levels and atherosclerosis. They state that hyperuricemia might have a protective role (antioxidant) or it could be a primary cause (pro-oxidant) in atherosclerosis (57). Protective role of uric acid has been established on animal model. Intraperitoneal injection of purine derivatives, caffeine or uric acid into male C57BL/6 mice significantly increased total glutathione (GSH) levels in the hippocampus (58). This study suggests that caffeine and uric acid induce neuronal GSH synthesis by promoting cysteine uptake, leading to neuroprotection (58). The clinical efficacy of uric acid as a protective antioxidant is currently under investigation in a Phase III trial because it was shown that co-administration of uric acid and recombinant tissue plasminogen activator provides synergistic neuroprotection in experimental thromboembolic models and protection of oxidative stress in patients with acute stroke (59,60).
It can be concluded that hyperuricemia is not directly responsible for vascular injury and increased risk for cerebrovascular disease but it simply represent a surrogate marker for high levels of damaging oxidative stress associated with increased xanthine oxidase activity (11). Indeed, hyperuricaemia is a significant predictor of disease state and progression of chronic heart failure (61). The assessment of UA is widely available at low cost, which may be an advantage for widespread determination of this marker. As it is preferably for biomarkers to fulfill the information for the evaluation of disease severity, prognosis and treatment (62) it is evidenced that uric acid determination is reasonable for heart failure. Manzano et al. have developed a novel risk model for elderly patients with heart failure which includes uric acid and left-atrial dimension and provide a reliable estimate of death or hospital admission rates over a 2-year follow-up period (63). This might help to identify higher risk patients who may benefit from more intensive treatment.
SUA and hypertension
Hypertension and increased SUA additionally complicate the controversies of uric acid role in cardiovascular events and the factors involved in pathogenesis of atherosclerotic consequences. Many clinical studies implicate on causative role of uric acid in hypertension (8,64). There are at least two aspects of interrelationships between hypertension and uric acid, molecular mechanisms and elimination pathways of urates.
For presentation of the first aspect we have reviewed a few clinical trials bellow. Randomized, double-blind, placebo-controlled, crossover trial involving 30 adolescents who had newly diagnosed, essential hypertension and SUA > 360 mmol/L and who were treated with allopurinol, resulted in reduction of blood pressure (65). Close association of new-onset essential hypertension in children; suggest that lowering of uric acid can lower blood pressure in some patients (66). SUA impacts blood pressure in pediatric hemodialysis patients, independent of volume, nutritional and weight status (67). The study on the Polish patients which was performed to evaluate the influence of allopurinol on blood pressure and aortic compliance in patients with arterial hypertension depending on hypotensive therapy with angiotensin-converting enzyme inhibitor (ACE-I) or thiazide diuretic, showed that allopurinol does not produce additional antihypertensive effects in patients with treated arterial hypertension (68). For understanding the mechanisms of uric acid roles either as a predictor or consequence, animal models has been investigated. In animal models it has been shown that induced mild hyperuricemia may contribute to endothelial dysfunction and reduction of nitric oxide (NO) levels (69). Uric acid causes hypertension in a rat model through the activation of the renin-angiotensin system, down regulation of nitric oxide, and induction of endothelial dysfunction and vascular smooth muscle proliferation (64). Some other studies showed that SUA protects against progression of hypertension in spontaneously hypertensive rats (70). Similarly to the vascular injury, understanding of the paradox should be pointed to metabolic pathway of uric acid production through xantine oxidase (1), as well as the differences between acting of uric acid inside the cells or in the extracellular milieu (71). We have already mentioned previously that uric acid acts as an antioxidant in extracellular fluid, while in cellular matrix exerts prooxidative effects (8,54).
It is well known how the impaired kidney function is involved in mechanisms of hypertension. The elimination of urate also depends on kidney function. The recent study on a large Korean cohort which determinate renal function and SUA levels showed that glomerular filtration rate negatively correlated with blood pressures, uric acid, fasting plasma glucose, lipid status and C-reactive protein and uric acid was found to be an independent factor associated with glomerular filtration rate (72).
2645 Greece systolic heart failure patients were involved in the Beta-Blocker Evaluation of Survival Trial (73). The results showed that hyperuricaemia might predict poor outcomes primary as a marker of increased xanthine oxidase activity. It confirmed significant association with poor outcomes in heart failure patients without chronic kidney disease (CKD) but not in those with CKD as a consequence of decreased renal excretion of uric acid (73).
Uric acid is primarily excreted via the urine. That excretion is dependent on a number of urate transporters, including urate transporter 1 (URAT1), organic ion transporters (OAT1 and OAT3) and ATP-dependent urate export transporters (MRP4) (74). Urate balance depends on those transporters activities. Transporter URAT 1 is responsible for the reabsorption of urate, which is normally 90% reabsorbed after glomerular filtration (75). When we are considering the influence of uric acid on hypertension it should be pointed to all factors which might be involved. Kidney function, functionality of the transporters, genetic polymorphisms of the transporters, factors of the oxidative stress have to be included in evaluation of interrelationships between hyperuricemia and hypertension. Impaired kidney function is responsible for many mechanisms involved in pathogenesis of hypertension, so the elevation of SUA might be one of them.
SUA and metabolic syndrome
It is well known that nephrolithiasis of uric acid origin is significantly more common among patients with the MetS, obesity and type 2 diabetes (9). Prevalence of metabolic abnormalities in Brazilian people with metabolic syndrome was associated with concentration of uric acid. After Roux-en-Y gastric bypass and consequently weight loss uric acid concentration were reduced (76). Lower urine pH was associated with an increase in waist circumference in over than thousand Japanese men (77). Waist circumference is a key feature for the metabolic syndrome and HOMA-R as an index of insulin resistance. Lower urine pH was also associated with an increase in BMI, and an increase in serum uric acid (77). SUA levels were significantly and negatively correlated with HDL-C in Korean males, but not in females but SUA levels increased significantly with an increasing number of MetS components and abdominal obesity in both genders (78). Elevated levels of uric acid in obese children, compared with lean subjects, at the prepubertal period, seems to be an early metabolic alteration that is associated with other features of insulin resistance syndrome (79). This might be explain with the presence of increased intracellular adenosine (uric acid precursor), a derivative of higher AMP concentrations due to increased synthesis of fatty acid-acyl-CoA in peripheral tissues (79), and the authors propose that in early obesity increased levels of uric acid are related to elevated plasma fatty acids. Molecular base for explanations of the interrelationship between hyperuricemia and MetS, we might also provide from high dietary carbohydrates intake especially for fructose or sucrose intake. On the other hand the animal model experiments showed that fructose feed rats vs. glucose feed rats, had higher body weight, blood pressure, and concentrations of insulin, uric acid and triglycerides (80).
Fructose enters hepatocytes and other cells (human and animal), where it is completely metabolized by fructokinase with the consumption of ATP, without negative regulatory mechanism to prevent the depletion of ATP like is in glucose metabolism. Lactic acid and uric acid are than generated in the process (81). Lowering of the urine pH is than the consequence of the organic acids accumulation. Metabolic fates of the fructose in the liver than increases the biosynthesis of endogenous triglyceride, VLDL excretion and LDL overproduction (82).
This also was confirmed in the clinical studies because it was shown that the reduction in serum uric acid correlated directly with the decrease in triglyceride levels (72). Any factor that may increase biosynthetic pathway in which ATP is consumed and adenilate is produces might be involved in overproduction of uric acid.
Severe and mild hyperuricemia have been recognized as an independent risk factor for endothelial dysfunction,flow-mediated vasodilatation of the brachial artery in subjects without MetS, whereas only severe hyperuricemia (but not mild hyperuricemia) appeared to exacerbate endothelial dysfunction in similar subjects with MetS in middle-aged healthy Japanese men (83).
The experiments on mice with targeted disruption of endothelial nitric oxide synthase (eNOS) showed that mice are hypertensive and insulin resistant. We have explained previously that uric acid may inhibit endothelial function by inhibition of NO-function under conditions of oxidative stress (37).
Measurement of uric acid concentration
Uric acid concentration might be measured in serum, plasma, urine (12) and in exhaled breath condensate (84). Determination of uric acid concentration includes phosphotungistic acid methods (PTA), uricase methods (in wide usage), high-performance liquid chromatography methods (reference methods), dry chemistry systems (12) and biosensor methods (85). PTA methods interfere with many endogenous and exogenous compounds. More specific and very low cost (less than 10 eurocents per analysis) are enzymatic methods with bacterial enzyme uricase, uricase- peroxidase standard method. Ascorbic acid and bilirubin interfere with urate determination, so the use of ascorbat oxidase and aminophenason is necessary (12). Up-to-date biosensor methods can also be used as a low-cost alternative to conventional methods (85).
Prior to determination of urate in urine, alkalinization of urine might be necessary, because of urate crystallize at pH lower than 5.75 (86). It is evidenced that uric acid concentrations are significantly associated with BMI, gender, glomerular filtration rate and hypertension (61). Many additional factors, including exercise (87), diet, drugs, and state of hydration, may result in transient fluctuations of uric acid levels (86). According to that the reference intervals for uric acid aren’t equable, and there is no clear limit for hyperuricemia. So it is very important to document well the strategy and the individuals included in the reference interval (88).
Conclusions
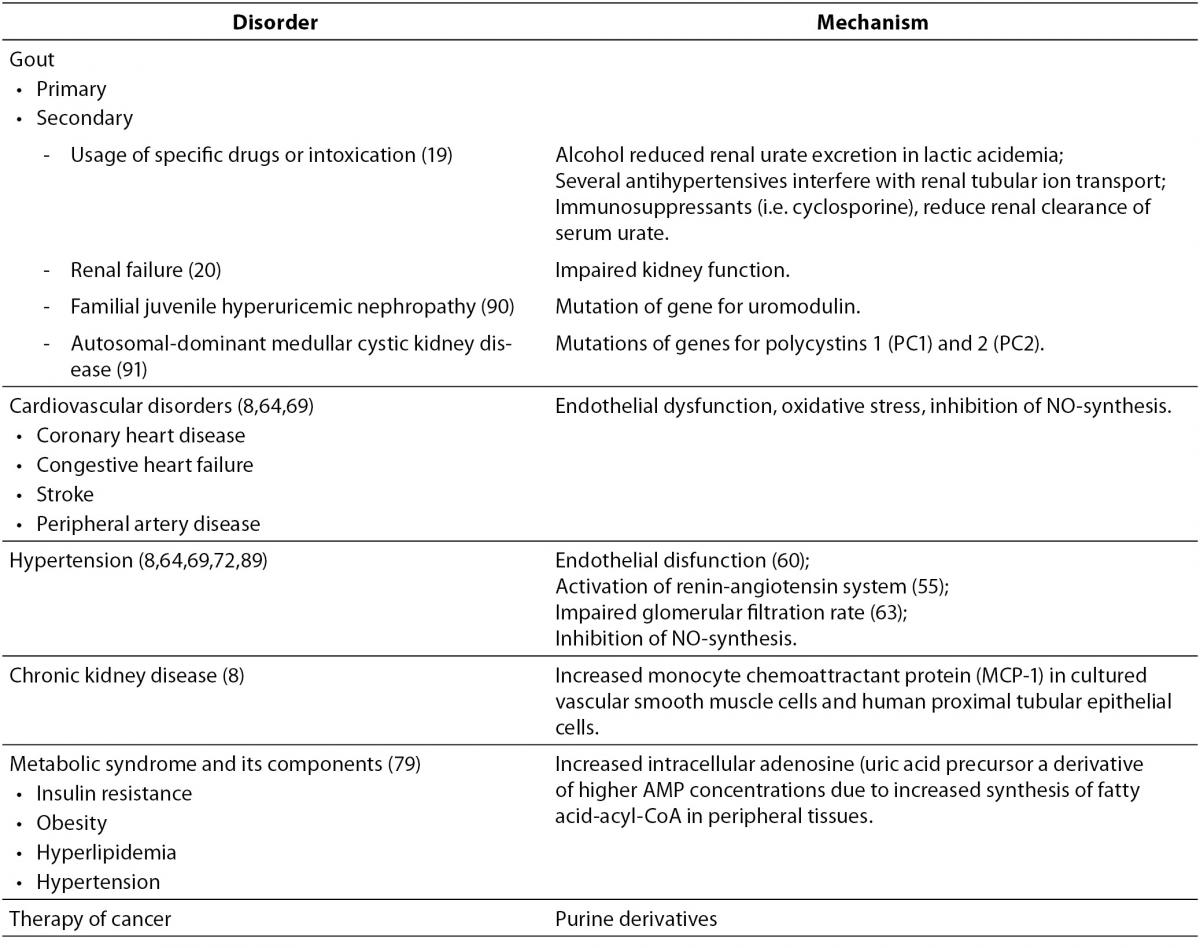
Instead of its controversial role in pathogenesis of different disorders, uric acid may play an important role in diagnosis, prognostic and therapy monitoring. SUA is a good indicator of the oxidative stress which is involved in pathobiochemisty of cardiovascular disorders, obesity, impaired glucose tolerance, hypertension and hyperlipidemias (Table 3). Hyperuricemia is not directly responsible for vascular injury and increased risk for cardiovascular or cerebrovascular disease but simply represents a surrogate marker for high levels of damaging oxidative stress associated with increased xanthine oxidase activity. Indeed, hyperuricaemia is a significant predictor of disease state and progression of chronic heart failure. The assessment of UA is widely available at low cost, which may be an advantage for widespread determination of this marker.
Table 3. Disorders and mechanisms involved in hyperuricemia.
Uric acid is not only recognized as a marker of oxidative stress. It also has a protective role and acts an antioxidant which is involved in clinical investigations of therapy for cerebrovascular disorders in combination with the anticoagulants.