Introduction
Cytomegalovirus (CMV) is a ubiquitous virus from the Herpesviridae family (1,2). The human CMV genome consists of a doublestranded DNA with approximately 230,000 bp (3). CMV is the most common cause of congenital infection in the developed world, occurring in ~1% of all liveborns (2,4,5). In 85-95% of newborns CMV infection runs asymptomatic, while in others it is presented with intrauterine growth restriction, microcephaly, hearing impairment, thrombocytopenia, anemia, jaundice, petechial rash, hepatosplenomegaly and central nervous system damage (1,2,4-6). Infants with symptomatic congenital CMV infection often have dramatic presentations, with a mortality rate of 10-30% (1,2,4,6). Infection in fetus/newborn can occur during primo-infection of mother, but also during reactivation of chronic latent infection and as a consequence of reinfection of seropositive mother (2,4). Infants born to mothers with primary CMV infection during pregnancy are more likely to have symptoms at birth (1). Treatment with ganciclovir/valganciclovir has to be considered for all children with symptomatic infection/disease, preliminary data have shown ganciclovir is effective in the treatment of symptomatic congenital CMV infection (2,5).
Alpha 1-antitrypsin (A1AT) deficiency is considered as the most common genetic liver disease in children (7). The gene encoding for A1AT is mapped on the chromosome 14 (8-10). A1AT, also referred to as alpha 1-proteinase (or protease) inhibitor (alpha 1-PI) is a 52 kDa, glycoprotein mostly secreted by hepatocytes and, to a lesser extent, by lung epithelial cells and phagocytes (8-11). It belongs to the family of serpins (serine protease inhibitors) and primarily neutralizes excessive proteolytic activity of neutrophil elastase. The altered conformation of the proteins encoded by the mutated alleles makes them prone to intensive intracellular accumulation or/and degradation in hepatocytes. A1AT deficiency is characterized by reduced serum A1AT level and/or impairment of A1AT protein. It is a common cause of neonatal cholestasis, hepatitis in neonates, childhood and adulthood and emphysema with or without hepatitis in adulthood (11-13). Numerous genetic variants of A1AT have been noted (14). Majority (95%) of numerous alleles of the A1AT gene, are termed as functional, reflecting their association with the synthesis if functional A1AT protein, while the remaining 5% are jointly named deficiency associated alleles. According to the frequency, they are classified as common (Z and S) and rare (Mmalton, null alleles etc.) (10,15). The most common phenotype is PiMM and is associated with normal levels of A1AT in serum. The two most common deficiency alleles are S and Z with S allele producing moderately low and Z allele very low levels of A1AT (8-10). In PiZZ phenotype serum A1AT concentration is characteristically reduced to 15% and in PiSS phenotype to 60% of normal levels. However, A1AT concentration can raise many folds upon inflammation, thus clinical manifestations among A1AT deficiency patients are under strong influence of environmental disease modifiers. An amount below the serum protective threshold increases risk for emphysema (16). For adults, the recommended consensus reference interval is 0.9 to 2.0 g/L for individuals with PiMM phenotype, with a median of approximately 1.3 g/L (14). AAT concentrations in PiZZ genotypes usually are 0.1 to 0.5 g/L (14). The concentrations and the variants are codominant; therefore an individual with PiMZ, for example would have a concentration of approximately 58% of the normal PiMM concentration [(100 + 15) / 2] (14). Neonates have higher concentrations, possibly because of maternal estrogen (14). AAT levels in children are lower than those in the adult population (17). Individuals with concentrations below 0.70 g/L should be phenotyped or genotyped, especially if they or family members have disease that may be associated with A1AT deficiency (14).
The production of defective A1AT protein leads to a decreased protective activity in the blood and lungs on one hand, while deposition of excessive abnormal A1AT protein in liver cells results in hepatocyte damage, on the other. The clinical spectrum of liver disease is very diverse; it ranges from the accidentally detected increased levels of transaminases through to severe infant cholestasis that can progress to cirrhosis. A1AT deficiency clinical manifestations develop as the result of interplay between mutations in A1AT gene and numerous inherited and acquired factors (i.e. smoking, air pollution, comorbidities, especially viral infections) (16,18).
According to available literature sources the current approach to specific therapy for individuals with lung disease related to A1AT deficiency involves administration of the normal A1AT protein (8). A1AT augmentatation is recommended only in affected patients functionally confirmed emphysema and homozygous presence of Z or rare allele, or their heterozygous combination. For those with no emphysema developed, the supportive therapy and preventive measures are considered sufficient. On the contrary, if the liver is affected, augmentation is not applicable and may even be deleterious. Nevertheless, effective supportive measures are included in therapeutic regimen intended for patients with liver damage (18). Therapeutic measures include nonspecific or supportive measures for clinical manifestations of liver and lung disease (8). In severe cases of liver or lung disease, transplantation has been used successfully in selected cases (8).
Both entities, congenital CMV infection and A1AT deficiency, are separately well described, but if they occur simultaneously their management can produce diagnostic and treatment challenges. Recently, case report concerning difficulties in misdiagnosing has been released (19). In this paper we describe a case of a male infant with unusually severe clinical course of congenital CMV infection presented by signs of early neonatal liver failure. Beside, the clinical manifestations of CMV infection in newborns include liver damage, unusually difficult clinical signs lead to assumption the additional liver damage exists. Raised suspicion on the underlying liver disease was verified by diagnosing A1AT deficiency. Clinical Hospital Centre Sestre milosrdnice (CHCSM) Ethics Committee approval has been obtained for writing this article.
Case report
A male two-month old infant was admitted for the first time to the Department of Paediatric Gastroenterology and Hepatology, CHCSM, for liver diagnostic work-up under suspicion of a congenital liver disease underlying severe clinical presentation of previously established CMV infection. The patient and his family members were Caucasians. The patient’s sister (5.5 years) suffered from spondylo-meta-epiphyseal dysplasia, and other family members were healthy. The patient was born from the second pregnancy and normal delivery, body weight (BW) 2910 g, body length (BL) 49 cm and APGAR 10/10. On the second day of life he presented with jaundice and petechial haemorrhages of the skin and was admitted to the Neonatology Department in another hospital. Laboratory tests revealed hypoglycaemia, thrombocytopenia, conjugated hyperbilirubinemia, hypoalbuminemia, increased activities of aspartate aminotransferase and alanine aminotransferase and significantly prolonged prothrombine time, whereas the activity of the gamma-glutamyltransferase was in the reference interval (Table 1). The multisliced computerized tomography (MSCT) (Siemens Somatom Emotion 2-sliced, Germany) revealed a choledochal cyst type I or II. Abdominal magnetic resonance imaging (MRI) (Siemens MAGNETOM Harmony 1T, Germany) revealed more voluminous cholecyst, medially elongated, and no other formations of the cystic contents. Intrahepatic bile ducts were not dilated, and extrahepatic bile ducts have failed to show up. Brain ultrasound (USD) (Siemens Sonoline G 40 TM) and Hearing Screening (OAE) were normal.
Table 1. Laboratory tests during follow-up period.
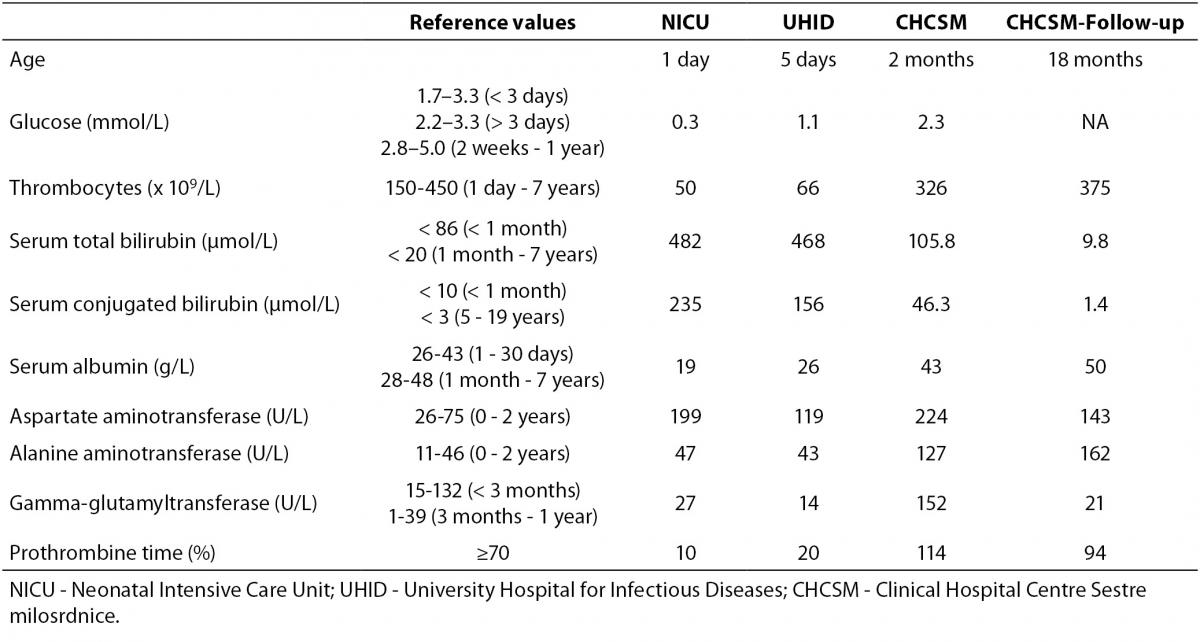
Augmentation [fresh frozen plasma (FFP) 15 mL/kg and 5% albumin 5 mL/kg] and antimicrobial therapy comprising ampicilin (100 mg/kg) and gentamycin (3 mg/kg) combination were initiated, accompanied with combined parenteral and enteral feeding due to a poorly tolerated oral intake of amino-acid based infant formula. Because the findings were suggestive of perinatal liver failure the patient was transferred to the Neonatal Intensive Care Unit (NICU), and later the same day moved to the University Hospital for Infectious Diseases (UHID) due to positive PCR for CMV DNA (CMV real-time PCR assay; Cepheid, CA, USA), both in blood (452 copies/mL) and urine (393 4210 copies/mL), but not in the cerebrospinal fluid. Samples for PCR for CMV DNA were taken during hospitalisation in NICU, and laboratory diagnostics performed at UHID. Upon admission to UHID parenteral treatment with ganciclovir along with symptomatic and supportive measures was introduced. Initially, despite daily transfusions of FFP (20 mL/kg/day prepared by standard procedures of responsible manufacturer, Croatian Institute for Transfusion Medicine), vitamin K (0.03 mg/kg/day), prothrombin complex concentrate, a mixture of clotting factors II, VII, IX, X and protein C and S (Octaplex, Octapharma Austria) at a dose of 4 mL (100 Units) during four consecutive days and antithrombin III (Antithrombin III Immuno, Baxter, USA) at a dose of 300 Units/daily (six doses cumulatively), coagulation parameters remained low, although visible bleeding was not recorded. Conjugated hyperbilirubinemia persisted, and at discharge serum total bilirubin was 205.8, direct 78 μmol/L. Only after four weeks (5th week of life) a graduate normalization of coagulation factors and platelets occurred. Hospitalization was complicated by catheter-related sepsis caused by Methicillin-resistant Staphylococcus aureus (MRSA), which has been treated with vancomycin (45 mg/kg) for 14 days with good response, and mild flow rotavirus gastroenteritis (real-time PCR method) treated with standard hypoosmolar oral rehydration solution. Parenteral treatment with ganciclovir (at a dose of 2 x 6 mg/kg/daily) was carried out on daily basis during 3 weeks and continued with oral treatment with valganciclovir (at a dose of 2 x 18 mg/kg/day) until CMV PCR DNA analysis from blood negativization at the age of 3 months. Oral intake (standard infant formula) was gradually introduced and increased with initially good start in weight gain, but at the end weight become stagnant. Psychomotor development was satisfactory. Presence of the infection with other hepatotrophic viruses [Hepatitis A virus (HAV), Hepatitis B virus (HBV), Hepatitis C virus (HCV), Epstein–Barr virus (EBV)] was tested; Anti-HAV total (IgM/IgG), Anti-HAV IgM, HBsAg, anti-HBs, total anti-HBc, IgM anti-HBc, HBeAg, anti-HBe, DiaSorin, Italy; HCV Ag-Ab, Bio-Rad, France; HCV confirmation test immunoblot/western blot, Innogenetics, Belgium; however only positive was PCR for CMV DNA. The infant was vaccinated with ENGERIX-B [Hepatitis B Vaccine (Recombinant)] GlaxoSmithKline, while vaccine against tuberculosis BCG was not administered.
At the age of 2 months the patient was transferred to the Department of Paediatric Gastroenterology and Hepatology, CHCSM for further liver work-up. At admission was in good general condition, 54.5 cm length [13th centile (c)], 4200 g weight (11th c; BMI 14.4 kg/m2), and head circumference was 40 cm (76th c). Skin and sclera were icteric, with no signs of rash or bleeding, decreased fat tissue especially on the limbs, normal bone and joint systems. Milder abdominal distension with soft liver palpable for 2 cm and spleen palpable for 1 cm was present. There were no signs of peripheral oedema or deformity. Neurological status was mildly hypotonic, in accordance with age.
Laboratory testing revealed reduced A1AT serum concentration (0.2-0.3, reference values 0.9-2.0 g/L), which was an important cooperative factor apparently necessary for the development of unusually severe clinical presentation in the first days of life with signs of liver failure. Due to a decreased A1AT serum concentration, for the child and his parents A1AT genotyping was performed by using LightCycler® 480 II (Roche Diagnostics Deutschland GmbH, Mannheim, Germany) real-time PCR method (LightMix® Kit Alpha1-Antitrypsin (AAT) Pi*S and Pi*Z, TIBMOLBIOL GmbH, Berlin, Germany). Genotyping was followed and confirmed with phenotyping (isoelectric focusing method followed by immunofixation on precasted agarose gel; semi-automated method on HYDRASYS Focusing system, Sebia, France), as it is our standard protocol for low A1AT serum values. The combination of genotyping and quantification, with a reflex to phenotyping, is the optimal strategy for the laboratory evaluation of A1AT deficiency (20,21). Genotype/phenotype results were concordant to each other: PiZZ phenotype for the child and PiMZ phenotype for both parents (mother A1AT serum concentration was 0.73, and father 0.71 g/L). A1AT genotyping was in concordance with phenotypes. Sweat test for cystic fibrosis screening, faecal elastase as well as thyroid hormone levels were normal. Abdominal USD (Siemens Sonoline G 40 TM, Germany) was unremarkable. Technetium 99m-hepatic iminodiacetic acid (HIDA) (Siemens Symbia T Germany) scintigraphy confirmed significant intrahepatic cholestasis with normal gallbladder function and the preserved passage into the intestine. Electrocardiographic and heart ultrasound (Siemens Sonoline G 40 TM) finding was normal. There were no signs that would raise suspicion of Alagille syndrome (22).
Therapy with ursodeoxycholic acid (UDCA; 10 mg/kg/daily), fat-soluble vitamin supplementation and medium-chain triglycerides (MCT) oil in meals was introduced along with hypercaloric feeding. UDCA may significantly improve clinical status and liver test results in some children with liver disease associated with PiZZ A1AT deficiency (23). High doses of vitamin D3 and other liposolubile vitamins (A, E, K) reimbursement continued as well as therapy with valganciclovir till the age of 3 months when CMV PCR DNA analysis from blood was negative. Coagulation parameters were corrected by daily FFP transfusions, vitamin K, prothrombin complex and antithrombin III supplementation. There was no need for vitamin K parenteral application. Hypercaloric diet (120% of the average daily caloric requirement) with extensively hydrolysed infant formula due to the higher MCT concentration was introduced and well tolerated.
Early introduced antiviral therapy and persistent supportive therapy in the first weeks of life bridged severe coagulation disorder, as well as hypoalbuminemia which were accompanied by all the expected consequences. In the further course liver disease was biochemically characterized by signs of moderate cholestasis with slowly but completely normalized synthetic and metabolic function. Despite the above mentioned events, the infant`s progress, somatic and psychomotor, was satisfactory. Within CMV infection there were no signs of the central nervous system impairment.
During the entire hospitalisation the patient was in good general condition, afebrile, vigorously crying, interestedly observing the surroundings, with good appetite and normal stools. He was discharged at the age of three months with a satisfactory weight gain (4750 g; 15th c) as well as somatic and motoric progress. Despite the high serum concentrations of bile acids (267.8; reference values < 10 µmol/L) there were no signs of pruritus.
The clinical course in the following 15 months showed excellent physical condition with normal weight gain and psychomotor development. At home, he was fed with frequent meals of extensively hydrolysed infant formula thickened with rice cereal and MCT oil added. The introduction of normal food was started at the age of 6 months. Vitamin D3 and multivitamin drops with docosahexaenoic acid (DHA) mL was continued and UDCA slowly tapering in accordance with the normalization of the liver function tests. At the age of 18 months, laboratory testing revealed no signs of cholestasys with normal values of serum bilirubin levels, bile acids (2.3 µmol/L) and vitamin D3 (170; reference values ≥ 75 nmol/L) levels, but with persistent signs of moderate hepatocellular lesion (Table 1).
Discussion
There is limited number of similar cases in the literature. Related cases report either CMV infection or A1AT deficiency, however not presented in same patient (5,7,13). There has been report of comorbid presentation of A1AT deficiency and CMV infection in adult (24). Additionally, one reported case presented 2-month infant, PiMM phenotype with urinary CMV, but without signs of hepatic necrosis, infection or cirrhosis (25). In our hospital the first case of A1AT deficiency was described in 1977, and one severe of PiZZ phenotype in 2007 (13,26). It was not associated with any other disease, such in this exceptional case (13). In our hospital this is the first case of such comorbidity presentation. The presented case provides excellent example about successful overcoming obstacles in differential diagnosis of A1AT in neonates and infants, as suggested by previous review (18). A simultaneously coexisting CMV infection and A1AT deficiency triggered severe clinical presentation. In this unique case, the underlying A1AT deficiency was enhanced by CMV infection. A1AT concentration can raise many folds upon inflammation. Clinical manifestations among PiZZ phenotype patients are under strong influence of environmental disease modifiers. In certain high risk groups, CMV infection may be dangerous (6). A1AT deficiency might be a relevant feature that predisposed to the severe clinical course of congenital CMV infection in this patient and vice versa. A1AT plays an important role in modulating immunity, inflammation, proteostasis, apoptosis, etc. A1AT has anti-inflammatory activities independent of its protease inhibitor function. The most frequently noted classical triad of clinical manifestations of CMV infection is jaundice (62%), petechias (58%), and hepatosplenomegaly (50%) and CMV infection could be disease stimulant in patients with the PiZZ phenotype that already are at risk to develop liver injury (1,6,12). Collaborative work of Neonatology, Intensive Care Unit, Infectious Diseases and Gastroenterology Paediatric Departments as well as specialists of laboratory and transfusion medicine, microbiologists and radiologists was essential in managing and resolving this case. Multidisciplinary teamwork was imperative for adding value of this report. Beside routine laboratory tests and imaging procedures recommended diagnostic procedures that have to be incorporated into routine diagnostic investigation of a child presenting with liver disease are CMV real-time PCR assay and determination of A1AT phenotype (1,6,27).
Optimal therapeutic approach was demand to be resolved and discussed. Taking into consideration severe clinical presentation of disease, an extensive treatment was introduced. Due to intensive and thorough supportive treatment during hospitalisation, at discharge from hospital and during the follow-up period the infant progress was satisfactory. In case of A1AT deficiency, liver functions need to be followed with a possibility to perform liver transplantation if considered necessary. Extensive diagnostic work-up, treatment and teamwork were imperative to give value to this particular case.
Prevention of the progression of lung diseases (eg. asthma, respiratory infection) is the main goal in management of subjects with A1AT deficiency. In addition, exposure to tobacco and air pollution should be avoided; choice of professional career may facilitate the main goal. Furthermore, since the Z allele is relatively frequent in the Caucasian population (about 1 in 50), genetic counselling for individuals and families who have diagnosis of A1AT deficiency, particularly in complex cases is of great importance.
Conclusion
The paper describes challenges in diagnosis and treatment of congenital CMV infection and A1AT deficiency which, in case of comorbidity, can present with a severe clinical course. There was obvious a relation between CMV infection and A1AT deficiency observed in our case. Their coexistence produced the expression of severe clinical presentation that emphasized the importance of CMV real-time PCR assay and A1AT phenotyping as part of routine diagnostic evaluation when dealing with liver dysfunction in infancy. It remains a challenge for further researchers as well as clinicians to investigate possible diagnostic and treatment correlations between these two well-known diseases.
Acknowledgment
The authors would like to thank Prof. Arijana Pavelić for her writing assistance and language editing.